A kidney disease's genetic clues are uncovered

Researchers have uncovered new genetic clues to understanding IgA nephropathy (IgAN), or Berger's disease, an autoimmune kidney disease and a common cause of kidney failure. The findings are relevant to IgAN as well as other diseases with similar underlying molecular defects, such as inflammatory bowel disease and certain types of blood disease and cancer.
"Very little is known about the causes of IgAN, genetic or otherwise, so our discovery represents an important step toward developing better therapies for this disease," said lead author Krzysztof Kiryluk, MD, the Herbert Irving assistant professor of medicine at Columba University Medical Center (CUMC).
The study, conducted by researchers at CUMC and the University of Alabama at Birmingham (UAB) School of Medicine, was published last month in PLOS Genetics.
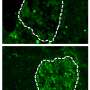
IgAN occurs when an antibody called immunoglobulin A (IgA) collects in the kidneys, causing inflammation of the glomeruli, the kidneys' filtering structures. Over time, the inflammation can hinder the kidneys' ability to filter waste from the blood. About half of patients with IgAN have progressive disease and eventually develop kidney failure. There is no cure for IgAN, but medications, along with blood pressure control, can slow disease progression.
The key molecular defect in people with IgAN is abnormal O-glycosylation of IgA antibodies. O-glycosylation—in which a sugar molecule attaches to an oxygen atom in the amino acid residue of a protein—plays a role in various physiologic processes. Studies of families have shown that problems in the O-glycosylation of IgA are common in people with IgAN and are largely genetic in origin, although the exact genes involved were unknown.
To identify genes linked to O-glycosylation problems in IgAN, Dr. Kiryluk and colleagues performed genome-wide association study (GWAS) of 2,633 people of European and East Asian ancestry, populations with high rates of the disease. All of the participants were analyzed for blood levels of galactose-deficient IgA1 (Gd-IgA1), a marker for IgAN, using a new high-throughput blood test developed by lead investigator of the study Jan Novak, PhD, associate professor of microbiology at UAB. A GWAS study of this kind had never been done before, because there was no way to efficiently measure the biomarker in such a large volume of patients.
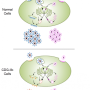
The researchers found that variations in two genes, C1GALT1 and C1GALT1C1, were significantly more common in people with high levels of the Gd-IgA1 marker. "The genes are found on different chromosomes, but they make proteins that interact to form an enzyme critical for the proper glycosylation of IgA molecules," said Dr. Kiryluk.
To confirm that C1GALT1 and C1GALT1C1 are involved in O-glycosylation, the researchers knocked down the two genes in cells in from IgAN patients and controls. Knocking down the genes increased production of the Gd-IgA1 marker in cells from both groups.
Variations in both genes, combined, accounted for about 7 percent of the overall variability in blood levels of Gd-IgA1 in the study population. "Since approximately 50 percent of variability in Gd-IgA1 levels is due to genetic factors, this means that about 43 percent of the genetic variability is still unexplained," said Dr. Kiryluk. "We started with a relatively small study population, so explaining 7 percent of variability between individuals with the disease was a good start. As we analyze more patients, we expect that we will find more genetic variants and can begin to piece together how these variants interact with environmental factors to cause disease." A GWAS study of some 10,000 patients is now underway.
More information: Krzysztof Kiryluk et al, GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway, PLOS Genetics (2017). DOI: 10.1371/journal.pgen.1006609