New study reveals how RNA splicing errors may spark the development of both ALS and a form of dementia
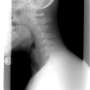
 is found in the nucleus of motor neurons (left), but it accumulates outside the nucleus in the neurons of patients with ALS and frontotemporal dementia who have a certain genetic mutation. Credit: Reed Lab/Harvard Medical School")
The most frequent genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)—rare and related neurological disorders marked by progressive deterioration of motor or cognitive abilities—may be due to errors in RNA splicing, an intermediary step for translating genetic instructions into functional proteins.
In a new study published June 13 in Cell Reports, researchers from Harvard Medical School show that toxic peptides produced by mutation of the C9ORF72 gene can prevent accurate assembly of the spliceosome—the molecular machine responsible for RNA splicing.
Genes affected by resulting splicing errors include those with mitochondrial, neuronal and gene expression functions. These processes have been previously linked to ALS and FTD, suggesting that restoring normal splicing activity may have potential as a therapeutic strategy for patients with ALS, FTD or both.
"Our findings indicate that the most prevalent mutation found in inherited ALS and FTD creates errors in spliceosome assembly," said senior study author Robin Reed, professor of cell biology at Harvard Medical School. "Since splicing is upstream of so many critical cellular functions, a better understanding of this mechanism could illuminate new approaches to help patients with these diseases, which currently have no effective treatments."
A specific mutation to the C9ORF72 gene accounts for around 25 percent of cases of FTD, where gradual loss of nerve cells in the frontal lobe of the brain leads to profound behavioral and cognitive deficits. Such mutations are also believed to fuel 30 to 40 percent of inherited forms of ALS, or Lou Gehrig's disease, a fatal disorder involving gradual loss of control over voluntary motor functions. Roughly one in five patients with ALS also develops FTD.
The C9ORF72 mutation causes the abnormal accumulation of many copies of a small, six-nucleotide long segment of DNA, which are processed by cells into messenger RNA—the molecules that carry instructions from DNA for the production of proteins. These extraneous messenger RNAs code for so-called dipeptide repeat proteins, two of which, GR and PR, have been found to be toxic in human, yeast and fruit fly cells.
Exactly how these peptides cause toxicity was, up until now, unclear, but previous studies have shown that they significantly increase the number of errors in splicing, the cellular process for editing raw messenger RNAs, which contain unnecessary segments that must be removed to accurately code for a protein.
Common thread
In their current study, Reed and colleagues discovered that the presence of GR and PR peptides cause a failure in the assembly of the spliceosome, the complex molecular machine that carries out RNA splicing. The team found that these toxic peptides strongly and specifically associate with a component of the spliceosome, known as U2 snRNP, causing it to aggregate in the cytoplasm of the cell instead of the nucleus, where it should normally be located.
The researchers observed this phenomenon in multiple cell types, including motor neurons derived from C9ORF72 patient stem cells. Additional analyses revealed that the failure of the spliceosome to assemble correctly led to splicing errors for numerous genes that depend on U2 snRNP for normal splicing.
These genes include many involved in the function of mitochondria, the power generators of the cell, whose malfunction has been previously linked to ALS. The team also found affected genes involved in neuronal structure and growth, and others that play roles in gene expression—cellular functions that have also been previously associated with ALS and FTD.
"It was striking how these peptides are so specific to U2 snRNP. No other cellular processes appeared to be affected, whereas splicing was completely blocked," Reed said. "When these peptides are expressed at high levels, they are completely toxic to the cell, but if they are produced at a low enough level, they can inhibit the splicing of genes that are U2-dependent, which may have some role in the development of disease."
It is currently unknown whether and how these mis-splicing events are involved in the development of ALS, FTD or other motor neuron diseases in human patients. While C9ORF72 mutations account for the majority of inherited forms of ALS and FTD, several other genes have also been implicated. In addition, more than 90 percent of ALS cases are sporadic, with no known genetic cause.
Mis-splicing, however, has been previously implicated in both inherited and sporadic ALS, FTD, as well as in spinal muscular atrophy, another motor neuron disease that the Reed lab previously showed shares biochemical pathways with ALS, suggesting that the process is a promising target for future therapeutic development.
"What we are finding is that disruptions in RNA splicing appear to be a common thread linking these motor neuron disorders," Reed said. "Much more research is needed, but if we could correct splicing errors with so-called splicing modulator compounds, we could prevent disruptions downstream at sites such as mitochondria, neuronal axons, or the neuromuscular junction, which may have efficacy for the treatment of ALS and FTD."