Genome therapy could lead to new treatment for life-threatening blood disorders

Genome therapy with beneficial natural mutation could lead to new treatment for life-threatening blood disorders
By introducing a beneficial natural mutation into blood cells using the gene-editing technique CRISPR, a UNSW Sydney-led team of scientists has been able to switch on production of foetal haemoglobin - an advance that could eventually lead to a cure for sickle cell anaemia and other blood disorders.
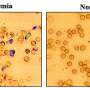
People with thalassaemia or sickle cell anaemia have damaged adult haemoglobin - the vital molecule that picks up oxygen in the lungs and transports it around the body - and they require life-long treatment with blood transfusions and medication.
However, people with these diseases who also carry the beneficial natural mutation - known as British-198 - have reduced symptoms, because the mutation switches on the foetal haemoglobin gene that is normally turned off after birth.
The extra foetal haemoglobin in their blood, which has a very strong affinity for oxygen, does the work of the defective adult haemoglobin.
"With CRISPR gene-editing we can now precisely cut and alter single genes within our vast genome," says study senior author and UNSW molecular biologist Professor Merlin Crossley.
"Our laboratory has shown that introducing the beneficial mutation British-198 into blood cells using this technology substantially boosts their production of foetal haemoglobin.
"Because this mutation already exists in nature and is benign, this 'organic gene therapy' approach should be effective and safe to use to treat, and possibly cure, serious blood disorders. However, more research is still needed before it can be tested in people," he says.
The study by scientists from UNSW, the Japanese Red Cross Society and the RIKEN BioResource Centre in Japan, is published in the journal Blood.
The beneficial British-198 mutation, which was first identified in a large British family in 1974, involves a change in just a single letter of the genetic code.
Carriers of this mutation have foetal haemoglobins levels as high as 20 per cent of total haemoglobin, while most people's foetal haemoglobin levels fall to about 1 per cent of total haemoglobin after birth.
The researchers also discovered how this British-198 mutation works. They found it creates a new binding site for a protein called KLF1 that turns blood genes on.
Mutations affecting adult haemoglobin production are among the most common of all genetic variations, with about 5 per cent of the world's population carrying a defective gene.
"To turn the new gene editing approach into a therapy for blood disorders, the British-198 mutation would have to be introduced into blood-forming stem cells from the patient," says Professor Crossley.
"A large number of stem cells would have to be edited in order to repopulate the patients' blood with genetically enhanced cells."
More information: Beeke Wienert et al, KLF1 drives the expression of fetal hemoglobin in British HPFH, Blood (2017). DOI: 10.1182/blood-2017-02-767400