Stabilizing TREM2 – a potential strategy to combat Alzheimer's disease

A gene called triggering receptor expressed on myeloid cells 2, or TREM2, has been associated with numerous neurodegenerative diseases, such as Alzheimer's disease, Frontotemporal lobar degeneration, Parkinson's disease, and Nasu-Hakola disease. Recently, a rare mutation in the gene has been shown to increase the risk for developing Alzheimer's disease.
Independently from each other, two research groups have now revealed the molecular mechanism behind this mutation. Their research, published today in EMBO Molecular Medicine, sheds light on the role of TREM2 in normal brain function and suggests a new therapeutic target in Alzheimer's disease treatment.
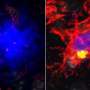
Alzheimer's disease, just like other neurodegenerative diseases, is characterized by the accumulation of specific protein aggregates in the brain. Specialized brain immune cells called microglia strive to counter this process by engulfing the toxic buildup. But as the brain ages, microglia eventually lose out and fail to rid all the damaging material.
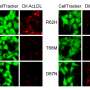
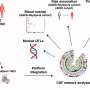
TREM2 is active on microglia and enables them to carry out their protective function. The protein spans the microglia cell membrane and uses its external region to detect dying cells or lipids associated with toxic protein aggregates. Subsequently, TREM2 is cut in two. The external part is shed from the protein and released, while the remaining part still present in the cell membrane is degraded. To better understand TREM2 function, the two research groups took a closer look at its cleavage. They were led by Christian Haass at the German Center for Neurodegenerative Diseases at the Ludwig-Maximilians-Universität in Munich, Germany, and Damian Crowther of AstraZeneca's IMED Neuroscience group in Cambridge, UK together with colleagues at the Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto and the Cambridge Institute for Medical Research, University of Cambridge, UK.
Using different technological approaches, both groups first determined the exact site of protein shedding and found it to be at amino acid 157. Amino acid 157 was no unknown. Only recently, researchers from China had uncovered that a mutation at this exact position, referred to as p.H157Y, increased the risk of Alzheimer's disease. Together, these observations indicate that protein cleavage is perturbed in the p.H157 mutant and that this alteration promotes disease development.
As a next step, Haass and Crowther's groups investigated the biochemical properties of the p.H157Y mutant protein more closely. They found that the mutant was cleaved more rapidly than a healthy version of the protein. "Our results provide a detailed molecular mechanism for how this rare mutation alters the function of TREM2 and hence facilitates the progression of Alzheimer's disease," said Crowther.
While most TREM2 mutations affect protein production, the mechanism behind p.H157Y is somewhat different. The p.H157Y mutation allows the protein to be correctly manufactured and transported to the microglia cell surface, but then it is cleaved too quickly. "The end result is the same. In both cases, there is too little full-length TREM protein on microglia," said Haass. "This suggests that stabilizing TREM2, by making it less susceptible to cleavage, may be a viable therapeutic strategy."
More information: TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer's disease-associated H157Y variant EMBO Molecular Medicine, DOI: 10.15252/emmm.201707673