Cystic fibrosis scientists discover abnormal response to lung infections

"For a very long time, there has been discussion about whether cystic fibrosis was a bacteria-infection problem, an inflammation problem, or an immune system problem," said Juan Ianowski, the lead author of the paper published today in the prestigious Nature Communications journal.
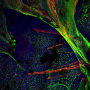
Ianowski, associate professor in the Department of Physiology at the University of Saskatchewan, and a team of 18 researchers have concluded that the genetic mutation that causes cystic fibrosis prevents normal secretion of airway surface liquid including mucus. Using a new imaging technique they developed at the Canadian Light Source, they determined that the production of airway surface liquid in response to bacteria is abnormal, and might lead to a cascade of infection and inflammation in lungs as the incurable disease progresses.
"Most patients are born with healthy lungs and as time passes, they start developing infections and inflammation. The infections are cleared and a new one occurs, and then there is an infection and it's cleared and a new one occurs. Eventually, the infections are permanent and the bacteria never leave the lungs," said Ianowski.
Cystic Fibrosis Canada estimates that one in every 3,600 children born in Canada has cystic fibrosis. It is the most common fatal genetic disease affecting Canadian children and young adults, according to the organization's website.
People with healthy lungs inhale between 5,000 and 14,000 germs every day. These germs are caught in their airways by a sticky mucus with antimicrobial properties that kills the germs. Tiny hairs called cilia then push the germs out of the respiratory tract and the body clears the mucus and the germs.
The hypothesis that people with cystic fibrosis do not have normal production of mucus in response to inhalation of pathogens was developed decades ago, but until now, there was no way to test it.
Ianowski and his team spent seven years developing the synchrotron-based method needed to analyze the production of mucus in a living animal. The normal layer of mucus is as thin as 80 microns. That's thinner than a strand of hair.
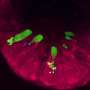
At the Canadian Light Source, they imaged the normal lungs of pigs focusing on the liquid layer, measuring it and analyzing its production. They also were able to image how the liquid in the airway reacts when bacteria are introduced. Then, they repeated the process with pigs who modeled cystic fibrosis.
"We discovered and showed for the first time ever that the normal airway response to inhalation of pathogens is producing liquid. This is a process that depends on CFTR (cystic fibrosis transmembrane conductance regulator) expression. So if you don't have normal CFTR, it doesn't work. In CF pigs, this response is absent," explained Ianowski.
More information: Xiaojie Luan et al. Cystic fibrosis swine fail to secrete airway surface liquid in response to inhalation of pathogens, Nature Communications (2017). DOI: 10.1038/s41467-017-00835-7