Mistakes in how proteins of the ear are built contribute to early hearing loss

From the moment a baby is born, they are put through a battery of screenings to test for all sorts of characteristics, which includes the sense of hearing. One in 500 newborns fail their newborn hearing screens and are diagnosed with hearing loss, making this the most prevalent sensory deficit in humans. About half of these cases of early hearing loss in developed countries have an identifiable genetic cause, with mutations in over 100 different genes identified so far.
Mutations in the majority of these genes result in isolated hearing loss (meaning not part of a more complex hearing loss syndrome such as Usher Syndrome). These cases are called non-syndromic sensorineural hearing loss, or SNHL, where an abnormal inner ear function is the only diagnostic feature. Despite the large number of identified hearing loss genes, the cause of inherited hearing loss remains a mystery in more than half the children.
A trio of researchers from the Perelman School of Medicine at the University of Pennsylvania and Children's Hospital of Philadelphia (CHOP) found mutations in a master-switch protein called Epithelial Splicing Regulatory Protein 1 (ESRP1) in individuals with SNHL. This research is published this week in Developmental Cell. In general, what connects most of the unexplained hearing-loss cases is that protein building in the cochlea during development goes awry. The cochlea has the all-important job of transforming mechanical energy in the form of sound waves into electrical signals that run along auditory nerves to the brain.
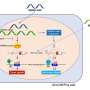
Overall, ESRP1 determines how RNAs expressed in epithelial tissues are spliced together. This is achieved by splicing different exons (the sequence of DNA that codes for proteins) together in alternative ways to produce more than one messenger RNA (mRNA) from the same gene. These mRNAs go on to make different versions of encoded proteins. Co-senior authors Doug Epstein, PhD, a professor of Genetics, and Russ Carstens, MD, an associate professor of Renal-Electrolyte and Hypertension, worked with Ian Krantz, MD, a professor of Pediatrics, and a family he cares for at the pediatric hearing clinic he directs at CHOP.
In the family, two out of the six children use cochlear implants for their hearing loss. The team sequenced the entire exome in the siblings and parents and found damaging mutations in ESRP1, with associated hearing loss. Using induced pluripotent stem cells made from affected and unaffected family members they showed that RNA splicing switches were restored when the ESRP1 mutation was corrected with CRISPR-CAS9 gene editing. "We were excited to see these results as they provided clear evidence that ESRP1 mutations were responsible for the splicing defects in the affected children" Epstein said. The gene editing experiments were performed by Penn co-authors Kiran Musunuru, MD, PhD, and postdoctoral fellow Chris McDermott-Roe.
Krantz and CHOP research intern, Ricky Tilton, MD, identified the ESRP1 gene mutations central to this family's case. "Beyond providing a long sought after answer for this family, this research is exciting as it implicates this critical molecular pathway with a developmental diagnosis in humans for the first time and helps shed light on a novel contributor to hearing loss that may lead to new approaches for therapeutics down the road," said Krantz, who is also director of Roberts Individualized Medical Genetics Center in the Roberts Collaborative for Genetics and Individualized Medicine at CHOP.
Mini Anatomy Lesson
"The act of hearing is based on hair cells in the ear that are like piano keys sensitive to vibrations at different pitches," Epstein said. According to John Germiller, MD, PhD, director of Clinical Research in the Division of Otolaryngology at CHOP, the two siblings in the family had a defect in the vestibular canals of their ears, but no obvious defect in the cochlea. In addition, loss of the Esrp1 gene in mice leads to changes in the shape of the inner ear that is very similar to the situation with the siblings.
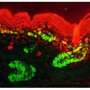
To determine how ESRP1 mutations cause hearing loss Alex Rohacek, a graduate student in the Epstein lab, evaluated embryos in which Esrp1 was deleted in a mouse model developed by the Carstens lab. The most striking finding in this part of the study was that formation of the stria vascularis was blocked. This set of cells is equivalent to the battery of the cochlea in that it provides hair cells with energy to transmit signals to the auditory nerve. These findings implicate mutations in ESRP1 as a cause of SNHL.
But how do alterations in RNA splicing result in hearing loss? Tom Bebee PhD, a postdoc in the Carstens lab and Rohacek compared RNA sequences from the cochleae of normal versus Esrp1 knockout mice. Working with a new algorithm developed by Yoseph Barash, PhD, an assistant professor of Genetics, the team made a list of differentially spliced genes. "We saw impaired expression and splicing of genes with essential roles in cochlea development and auditory function," Carstens said. At the top on the list was a Fibroblast growth factor receptor gene that (Fgfr2) that Carstens had used to identify ESRP1 in earlier research.
Because of altered Fgfr2 splicing, cells of the stria vascularis are missing in Esrp1-deficient mice, and possibly in humans. In the mice, formation of the stria vascularis was recovered by taking away a copy of Fgf9. The team says that this was one of the most compelling aspects of their study, because it speaks to the remarkable plasticity that exists in the genetic programs regulating inner ear development.
Although this line of research is far from contributing to changes in the clinic, the team now has a much better understanding of how RNA splicing is controlled in the ear. This knowledge opens up possibilities about treatment ideas that can be tested in animal models as a first step toward human trials.