December 22, 2017 report
Cancer cells that escape from senescence found to have an enhanced capacity to drive tumor growth

An international team of researchers has found that cancer cells that escape from senescence due to use of chemotherapy have an enhanced capacity to drive tumor growth. In their paper published in the journal Nature, the group describes their study that involved mouse models and cancer cells that escaped cell-division arrest after exposure to chemo drugs. Jan Paul Medema with the Cancer Center Amsterdam offers a News and Views piece on the work done by the team in the same journal issue.
In order to prevent damaged cells from dividing, resulting in the growth of abnormal tissue, cells have a built-in mechanism called senescence—cell division is arrested, preventing the cell from causing problems. Over the past several years, researchers have taken advantage of this mechanism to combat tumor cells—stopping cell division in senescent cells keeps the tumor from growing. Drugs have been developed and used as part of a chemotherapy regimen to treat cancers in this way. But now, it appears that there may be a side effect to such treatments—the researchers report tumors that escape senescence grow faster.
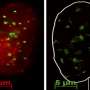
To learn more about what happens to tumor cells that survive senescence, the researchers started with the observation that signaling pathways in cells that have been forced into senescence were similar to those that have been seen in stem cells. To find out if there might be a connection, they created lymphoma mouse models with an induced state of cell-cycle arrest via the drug tamoxifen—stopping administration of the drug allowed the cells to exit senescence on-demand, allowing the researchers to watch what happened to the tumors. The researchers report that in nearly all cases, they found faster-growing tumors.
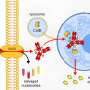
Upon taking a closer look at what was occurring, the researchers found that senescence in the cells was connected to the activation of the Wnt signaling pathway—a group of proteins that pass signals through cell surface receptors. Interestingly, they also found that if they added Wnt inhibitors after tamoxifen cessation, tumors grew slower after exiting senescence, suggesting that adding such inhibitors to chemotherapy mixtures might be coming in the future.
More information: Maja Milanovic et al. Senescence-associated reprogramming promotes cancer stemness, Nature (2017). DOI: 10.1038/nature25167
Abstract
Cellular senescence is a stress-responsive cell-cycle arrest program that terminates the further expansion of (pre-)malignant cells1,2. Key signalling components of the senescence machinery, such as p16INK4a, p21CIP1 and p53, as well as trimethylation of lysine 9 at histone H3 (H3K9me3), also operate as critical regulators of stem-cell functions (which are collectively termed 'stemness')3. In cancer cells, a gain of stemness may have profound implications for tumour aggressiveness and clinical outcome. Here we investigated whether chemotherapy-induced senescence could change stem-cell-related properties of malignant cells. Gene expression and functional analyses comparing senescent and non-senescent B-cell lymphomas from Eμ-Myc transgenic mice revealed substantial upregulation of an adult tissue stem-cell signature, activated Wnt signalling, and distinct stem-cell markers in senescence. Using genetically switchable models of senescence targeting H3K9me3 or p53 to mimic spontaneous escape from the arrested condition, we found that cells released from senescence re-entered the cell cycle with strongly enhanced and Wnt-dependent clonogenic growth potential compared to virtually identical populations that had been equally exposed to chemotherapy but had never been senescent. In vivo, these previously senescent cells presented with a much higher tumour initiation potential. Notably, the temporary enforcement of senescence in p53-regulatable models of acute lymphoblastic leukaemia and acute myeloid leukaemia was found to reprogram non-stem bulk leukaemia cells into self-renewing, leukaemia-initiating stem cells. Our data, which are further supported by consistent results in human cancer cell lines and primary samples of human haematological malignancies, reveal that senescence-associated stemness is an unexpected, cell-autonomous feature that exerts its detrimental, highly aggressive growth potential upon escape from cell-cycle blockade, and is enriched in relapse tumours. These findings have profound implications for cancer therapy, and provide new mechanistic insights into the plasticity of cancer cells.
© 2017 Phys.org