Defects on regulators of disease-causing proteins can cause neurological disease
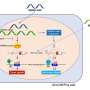
When the protein Ataxin1 accumulates in neurons it causes a neurological condition called spinocerebellar ataxia type 1 (SCA1), a disease characterized by progressive problems with balance. Ataxin1 accumulates because of a mutation that produces an abnormal, very long version of the protein that forms clumps inside neurons. However, studies on animal models have shown that accumulation of normal, regular-length Ataxin1 can lead to neurodegeneration similar to SCA1. In this case, the problem was a mutation in one of the genes that regulate Ataxin1 production, called Pumilio1, that resulted in overproduction of normal Ataxin1. Inspired by the findings in animal models, an international team of researchers explored the possibility that a similar phenomenon happened in people. They report in the journal Cell that mutations in human PUMILIO1 gene also cause conditions similar to SCA1. These findings provide a better understanding of the genetic basis of neurological diseases and suggest that identifying protein regulators would single out new candidates for disease-causing genes.
"In this study, we investigated the possibility that our findings in mice—that mutations in Ataxin1 regulator Pumilio1 cause a neurological disease similar to SCA1—would translate to people," said first author Dr. Vincenzo Gennarino, a postdoctoral fellow in Dr. Huda Zoghbi's lab at Baylor College of Medicine during the development of this work, and currently an assistant professor of genetics and development at Columbia University Medical Center.
The amount of a single protein can affect the manifestation of a disease
The researchers began by searching for patients with symptoms similar to those of SCA1 in large genomic databases around the world. They identified 15 patients with different mutations of PUMILIO1 gene, which they separated into two groups. In one group, the amount of PUMILIO1 protein was about half that detected in people without the condition. These patients presented with a severe form of the disease that had started early, between 5 months and 1 year of age. The children have seizures, ataxia, poor motor coordination and developmental delay. In the other group of patients, the amount of PUMILIO1 was 75 percent of that present in individuals without the condition. These patients presented with a milder form of ataxia that appeared in their 40s or 50s.
"This study, which is a nice example of the power of collaborations, taught us that mutations in the same gene can cause vastly different neurological problems based on how much they compromise the protein product," said corresponding author Dr. Huda Zoghbi, professor of molecular and human genetics and of pediatrics and neuroscience at Baylor and director of the Jan and Dan Duncan Neurological Research Institute. Zoghbi also is an investigator at the Howard Hughes Medical Institute.
"We found these findings very exciting," Gennarino said. "We learned that the characteristics of the disease vary according to the levels of PUMILIO1 protein. We used to think that modest changes in the levels of certain proteins, such as a 25 percent deficiency, had no detrimental effect on the individual. Here we found that this is not the case. This is an important finding from the clinical point of view. An individual born with a 25 percent deficiency of PUMILIO1 or perhaps other proteins may only develop symptoms later in life."
These findings also have implications for other neurodegenerative conditions such as Alzheimer's disease, amyotrophic lateral sclerosis, or Lou Gehrig's disease, and Parkinson's disease. Identifying the regulators of proteins involved in these diseases would provide a list of candidate genes potentially linked to the condition and open new venues for novel therapeutic strategies.
"We hope that this study encourages other researchers to look back at their own studies from the perspective of the regulators of protein synthesis or clearance," Gennarino said.
More information: Cell (2018). DOI: 10.1016/j.cell.2018.02.006