Researchers define target and mechanism of antibacterial drug fidaxomicin (Dificid)
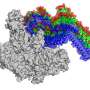
 binds to RNA polymerase (gray) at the base of the clamp, a part of RNA polymerase that swings open by 17 dgrees to allow RNA polymerase to bind to DNA and swings closed to allow RNA polymerase to hold onto DNA (open clamp in red at upper left; closed clamp in gray at upper left). Fidaxomicin locks the RNA polymerase clamp in the open position (red). Credit: Wei Lin and Richard H. Ebright, Rutgers University")
A team of Rutgers University and international scientists has determined the molecular target and mechanism of the antibacterial drug fidaxomicin (trade name Dificid).
Fidaxomicin was approved in 2011 for treatment of the CDC "urgent threat" bacterial pathogen Clostridium difficile (C. diff) and currently is one of two front-line drugs for treatment of C. diff.
Fidaxomicin also exhibits potent antibacterial activity against other CDC "serious threat" bacterial pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus (VRSA), and the tuberculosis bacterium, Mycobacterium tuberculosis. However, the low solubility and low systemic bioavailability of fidaxomicin have precluded use of fidaxomicin for treatment of MRSA, VRSA, and tuberculosis.
To design next-generation fidaxomicin derivatives with improved clinical activity against C. diff and useful clinical activity against MRSA, VRSA, and tuberculosis, it is essential to know how the drug binds to and inhibits its molecular target, bacterial RNA polymerase, the enzyme responsible for bacterial RNA synthesis.
In a paper published in Molecular Cell today, the researchers report results of cryo-electron microscopy (cryo-EM) and single molecule spectroscopy analyses showing how fidaxomicin binds to and inhibits bacterial RNA polymerase.
The researchers report a cryo-EM structure of fidaxomicin bound to Mycobacterium tuberculosis RNA polymerase at 3.5 Å resolution. The structure shows that fidaxomicin binds at the base of the RNA polymerase "clamp," a part of RNA polymerase that must swing open to allow RNA polymerase to bind to DNA and must swing closed to allow RNA polymerase to hold onto DNA. The structure further shows that fidaxomicin traps the RNA polymerase "clamp" in the open conformation.
The researchers also report results of single-molecule fluorescence spectroscopy experiments that confirm that fidaxomicin traps the RNA polymerase "clamp" in the open conformation and that define effects of fidaxomicin on the dynamics of clamp opening and closing.
The researchers show that fidaxomicin inhibits bacterial RNA polymerase through a binding site and mechanism that differ from those of rifamycins, another class of antibacterial drugs that target bacterial RNA polymerase. The finding that fidaxomicin inhibits bacterial RNA polymerase functions through a different, non-overlapping binding site and mechanism explains why fidaxomicin is able to kill bacterial pathogens resistant to rifamycins and why fidaxomicin is able to function additively when combined with rifamycins.
The new results enable rational, structure-based design of new, improved fidaxomicin derivatives with higher antibacterial potency, higher solubility, and higher systemic bioavailability. Based on the structure of fidaxomicin bound to its target, the researchers identified atoms of fidaxomicin that are not important for binding to the target and thus that can be modified without compromising the ability to bind to the target. The researchers then developed chemical procedures that allow selective attachment of new chemical groups at those atoms, including new chemical groups that can improve potency, solubility, or systemic bioavailability.
"The results set the stage for development of improved fidaxomicin derivatives, particularly improved fidaxomicin derivatives having the solubility and systemic bioavailability needed for treatment of systemic infections, such as MRSA and tuberculosis," said Ebright, Board of Governors Professor of Chemistry and Chemical Biology and Laboratory Director at the Waksman Institute of Microbiology at Rutgers, who led the research.
More information: Molecular Cell (2018). DOI: 10.1016/j.molcel.2018.02.026