Study identifies new molecular target for treating deadly lung disease IPF
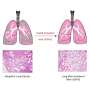
, extensive fibrosis and collagen deposits (shown in red) have extensively infiltrated the tissue. Researchers at Cincinnati Children's report in Cell Reports that loss of the gene Foxf1 in lung myofibroblasts increased the severity of IPF in mice. Credit: Cincinnati Children's")
Scientists searching for a therapy to stop the deadly and mostly untreatable lung disease, idiopathic pulmonary fibrosis (IPF), found a new molecular target that slows or stops the illness in preclinical laboratory tests.
A research team at Cincinnati Children's Hospital Medical Center reports its data in the journal Cell Reports. It found that a gene called FOXF1 inhibits the IPF disease process, which includes extensive scarring in lung connective tissues, hyper-production of harmful cells called myofibroblasts and excessive lung inflammation.
Their data show human lungs from IPF patients and mouse models of IPF lack FOXF1 in myofibroblasts. But cells lacking FOXF1 also exhibit overexpression of a related gene called FOXM1, which drives lung scarring and inflammation.
"The exact cause of IPF is unknown and effective treatments are needed. This study identifies a novel anti-fibrotic drug target that inhibits pulmonary fibrosis in our preclinical models," said lead investigator Tanya Kalin, MD, PhD, Division of Pulmonary Biology. "We are developing different therapeutic approaches and conducting preclinical tests to increase FOXF1 expression in the cells of lung connective tissues."
Although Kalin stressed that an estimated two to three years of additional laboratory study and development are needed before it's known if the data can be applied to clinical treatment, the research team is working on several potential therapeutic tools to boost FOXF1 levels in IPF. This includes testing a novel small-molecule compound that stabilizes FOXF1 and inhibits myofibroblasts in IPF. Researchers also are testing a nanoparticle-based gene delivery method to get FOXF1 into lungs with IPS.
Untangling a Mystery
FOXF1 is a transcription factor—a regulator gene that essentially tells other genes what to do, including what proteins to make and biological processes to help initiate or inhibit. The current study shows FOXF1 is supposed to act as a molecular switch for two other genes called cadherins (CDH2 and CDH11).
FOXF1 induces CDH2, but inhibits CDH11 to shut down IPF if something triggers it. Researchers performed extensive lab and genetic analyses on biopsied human lungs donated by IPF patients and mouse models of IPF to find out that when FOXF1 is repressed its sister gene FOXM1 increases and fuels the disease.
IPF's precise causes remain unknown, but genetic predisposition, smoking and other potential environmental factors are suspected. Also under investigation are the effects these factors may have on human epigenetics—how environment changes regulate the activity of our genes.
To induce and study the IPF disease process in laboratory mice, the rodents were administered a chemotherapy agent for treating certain cancers called bleomycin. The chemical is toxic to the respiratory system and triggered tissue damage and fibrotic scarring in the animals' lungs. Human IPF biopsy samples were also used in these studies.
According to the National Library of Medicine at the National Institutes of Health (NIH), IPF usually affects people between the ages of 50 and 70, but it can strike younger adults and children. About 100,000 people are affected in the United States, with an estimated 30,000 to 40,000 new cases diagnosed annually.
The most common signs and symptoms of the disease are shortness of breath and a persistent dry, hacking cough.