Scientists discover a mechanism of drug resistance in breast and ovarian cancer

There is a highly sophisticated way to treat some breast and ovarian cancers—a class of drugs called PARP inhibitors, designed to exploit the very defects that make tumors with certain mutations especially deadly. Yet this targeted approach to cancer therapy sometimes fails, and scientists have anxiously sought to understand why.
Now, research at The Rockefeller University offers insight into the biology behind this drug resistance and fresh hope for fighting it. Scientists in Titia de Lange's lab and their colleagues have discovered the molecular means by which some cancers caused by errors in the gene BRCA1 evade treatment by drugs custom-tailored to kill them.
Described in Nature on July 18, their work challenges previous assumptions about the mechanics by which these PARP inhibitors succeed or fail to help patients.
"This is a complete shift in our understanding of the mechanism that underlies this form of treatment for BRCA1 cancers," says de Lange, the Leon Hess Professor.
Their discovery helps to explain why some cancers respond to PARP inhibitors, while others do not—an insight that could ultimately be used to help improve treatments for patients.
A defect—and an opportunity
Experts predict that about 288,000 new cases of breast and ovarian cancer will be diagnosed this year. A substantial fraction of these cancers are caused by harmful errors in two of the most infamous constituents of the human genome: the genes known as BRCA1 and BRCA2. Mutations in BRCA1—the subject of the new research—have been estimated to give a woman a roughly 72 percent chance of developing breast cancer and a 44 percent chance of developing ovarian cancer by the age of 80.
Both genes are tumor suppressors, meaning they normally help keep the body cancer-free. They code for proteins that are important for properly repairing a DNA molecule that has been cut somewhere along its length—a mishap called a double-strand break, since it severs both strands of the DNA helix. Without the BRCA genes, the broken DNA isn't fixed properly, producing mutations that can lead to cancer.
In recent years, the development of new drugs called PARP inhibitors made it possible to turn those same genetic defects against the disease. The drugs prompt the formation of double-strand breaks; unable to properly repair these breaks, BRCA-deficient tumor cells die.
However, some tumors that should be vulnerable to PARP inhibitors don't respond. Scientists think this failure occurs for a number of reasons, and researchers in de Lange's lab homed in on one culprit linked to resistance in BRCA1 cancers in particular.
For nearly a decade, scientists had known that the loss of a protein called 53BP1 made it possible for BRCA1-deficient cells to overcome their inherent defect and properly repair double-strand breaks. This type of resistance can emerge during or after PARP inhibitor treatment when some tumor cells thrive after mutating to lose 53BP1. But it wasn't clear why losing this protein gives these cancer cells such a deadly advantage.
A different mechanism
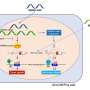
In order to prepare a broken DNA molecule for repair, one strand of the broken double-helix first needs to be trimmed back. It was assumed that 53BP1 prevents this pruning. Lose 53BP1, the thinking went, and BRCA1-deficient cells suddenly gain the ability to repair their DNA breaks.
In experiments, de Lange's team showed that 53BP1 does something different. Work by Zachary Mirman, a graduate student in the lab; Francisca Lottersberger, a former postdoc; and their colleagues found that 53BP1 instead helps to counteract the pruning process by rewriting sections of DNA cut from these loose strands.
In BRCA1-deficient cancers treated with PARP inhibitors, the rewriting function of 53BP1 leads to faulty repair of DNA and the death of the cancerous cells. Yet some of these cells manage to escape treatment by losing 53BP1. The work from de Lange's lab explains just how this change allows them to survive.
"In the big picture, this new insight into 53BP1's function and its role in drug resistance provides a foundation for advancements in PARP inhibitor therapy," Mirman says. These improvements could include the development of screening tests to see which tumors would best respond to PARP inhibitors, or to determine which other drugs should—or should not—be given along with them, the researchers say.
More information: Zachary Mirman et al, 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Polα-dependent fill-in, Nature (2018). DOI: 10.1038/s41586-018-0324-7