Drug identified that could reverse pulmonary arterial hypertension
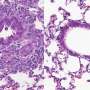
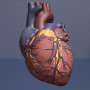
Scientists at Stanley Manne Children's Research Institute at Ann & Robert H. Lurie Children's Hospital of Chicago took a major step toward developing a new treatment for pulmonary arterial hypertension (PAH), a severe lung disease with a five-year survival rate of 50 percent. They identified a drug with a positive safety profile that inhibits a gene called HIF-2α, which they discovered earlier promotes the progressive thickening of the lung artery walls—a key feature of PAH called "vascular remodeling," which leads to right-sided heart failure, the main cause of death in PAH patients. Recently, they demonstrated in three clinically-relevant animal models that inhibiting HIF-2α with a compound results in reversal of established PAH, suppression of vascular remodeling and right heart failure, and increased survival. These findings were published in the American Journal of Respiratory Critical Care Medicine.
"We are thrilled to reach this critical stage in developing the first drug for pulmonary arterial hypertension that targets the mechanisms behind disease development," says lead author Zhiyu Dai, Ph.D., from the Manne Research Institute at Lurie Children's, who also is a Research Assistant Professor of Pediatrics at Northwestern University Feinberg School of Medicine. "We plan to complete preclinical testing of the new drug by the end of 2018 and launch a clinical trial in 2019."
In PAH, vascular remodeling causes high blood pressure in the lung arteries, interferes with the smooth flow of blood from the heart and ultimately results in right heart damage. In severe cases, the pulmonary arteries become completely blocked, leading to right heart failure and death. Currently, there is no effective therapy to reverse vascular remodeling and inhibit right heart failure in PAH. Available treatments mainly target vasoconstriction, or contraction of the lung arteries, achieving only modest improvements in PAH morbidity and mortality.
"It is exciting to see our research progressing from the bench to the bedside," says senior author and program director Youyang Zhao, Ph.D., from the Manne Research Institute at Lurie Children's and Professor of Pediatrics, Medicine and Pharmacology at Northwestern University Feinberg School of Medicine. "We started with creating a unique genetically modified mouse model that is the first to mimic how pulmonary arterial hypertension develops in patients. This helped us establish the potentially therapeutic benefits of inhibiting HIF-2α and the feasibility of doing this with a compound. Now we have a promising drug that we know is safe to test in a clinical trial. We are hopeful that it can reverse vascular remodeling in patients with this devastating disease and ultimately save lives."