Novel molecular target to prevent scarring of the lung blood vessels identified
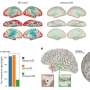
 for 24 hours shows co-localization of NEDD9 (red) and NKX2-5 (green), DAPI (blue), 63x. Credit: Bradley Maron, Brigham and Women's Hospital")
Pulmonary arterial hypertension, a severe form of cardiopulmonary disease in which the arteries that transport blood from the heart to the lungs become thickened, constricted, and scarred, is a disease for which there is no cure. Investigators from Brigham and Women's Hospital are unraveling the molecular mechanisms that may control PAH's development and progression in an effort toward finding treatments that can halt the disease's advancement.
In a paper published this week in Science Translational Medicine, researchers share results from a study that identifies the cancer protein NEDD9 as a critical player in disease development, with potential therapeutic implications for patients with PAH.
"Our work identifies as a novel molecular target to prevent fibrosis in lung arteries, which can lead to early right-sided heart failure and death," said corresponding author Bradley Maron, MD, a cardiologist and vascular biologist at BWH. "Our data provide an alternative mechanism that may account for the variability in fibrosis observed across subgroups of patients with PAH."
In PAH, fibrosis (scarring) is an important cause of damage to pulmonary arteries, which increases strain on the heart and leads to perpetual shortness of breath. In the past, researchers had theorized that one pathway acted as a "master switch" to globally control fibrosis but new evidence suggests that the development of fibrosis may be more complicated and more specific to different kinds of tissue. To better understand which genes and proteins may influence scarring specifically in the lung arteries, the Brigham research team used network medicine—a method of mapping relationships between proteins, pathways and more. Through this approach, the researchers discovered that the cancer protein NEDD9 is a critical mediator of vascular fibrosis in lung blood vessels.
They went on to identify a specific amino acid residue in NEDD9 that controls lung artery fibrosis. In patients with PAH, increased oxidant stress modifies this NEDD9 amino acid to cause lung artery fibrosis. The team also showed that the lung artery endothelium, previously regarded largely as a bystander in vascular fibrosis, contributes to this problem.
At the heart of the research team's findings is NEDD9, which their data suggest mediates fibrotic vascular remodeling. The authors note that this has potential therapeutic relevance for PAH patients. While a therapy that targets this protein has not yet been developed, because of its role in cancer, several studies have put NEDD9 forward as an important therapeutic target for future drug discovery efforts.
More information: A.O. Samokhin el al., "NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension," Science Translational Medicine (2018). stm.sciencemag.org/lookup/doi/ … scitranslmed.aap7294