September 24, 2018 weblog
How to edit your mitochondria

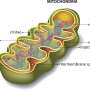
Mitochondrial genetic engineering is the adaptation of genetic engineering techniques to specific mitochondrial problems. Although it is not common to be born with severe mitochondrial issues, we will all eventually have to face these problems in one form or another, provided we live long enough.
Perhaps the most powerful tool currently available for controlling the code of the mitochondrial genome is gene-editing technology. The CRISPR/Cas9 system, in particular, has been used with great success in the nuclear genome. Unfortunately, there are several reasons why CRISPR is problematic for mitochondria. For one, there are not that many sites in the comparatively small mitochondrial genome where CRIPSR could be effectively brought to bear. Furthermore, there is no clearly defined mechanism for importing the guide RNA into mitochondria. These facts of life make the few published claims of any successful mito-CRISPR rather dubious.
A much more attractive option for editing mitochondria is to use zinc finger nucleases (ZFNs) engineered to cut double-stranded DNA at desired locations. ZFNs work in pairs—one part targets the offending nucleotide on one strand while the other part is designed to bind a specific number of nucleotides away on the opposite strand. If all goes well, the two halves will eventually dimerize and cleave the DNA.
One seeming roadblock is that splicing in the 'correct' nucleotide and then patching things back together is difficult because mammalian mitochondria don't do double-stranded repair very efficiently, if at all. The beauty of the ZFN approach is that one doesn't actually need to do a full edit and repair. Any mtDNA molecules with double-strand breaks are broken down and eliminated in short order by the endogenous nuclease activity of PolG, the mitochondrial DNA polymerase, and MGME1, a mitochondrial endonuclease involved in mtDNA replication. The cell then responds accordingly to bring mtDNA levels back into balance by upregulating nucleotide pools and printing new mtDNA. Rather then performing single-genome editing per se, one is actually editing the mitochondria at the population level to achieve a desired level of heteroplasmy between different (and not necessarily all bad) genomes.
Payam Gammage, from MRC Mitochondrial Biology Unit at the University of Cambridge, and his colleagues have just reported the first in vivo successes in using zinc finger nucleases for selectively editing away pathogenic mtDNAs in a heteroplasmic mammalian mitochondrial population. Writing in the Sept. 24th issue of Nature Medicine, they demonstrate the correction of a cardiac-specific mitochondrial disorder with concomitant therapeutic restoration of molecular and biochemical hallmarks to an undiseased state.
To do this, they used a mouse model that contains well-defined levels of heteroplasmy for the mitochondrial tRNA-encoding alanine. The notation for this variant reads m.5024C>T tRNA(Ala). Because the alanine tRNA is actually coded on the light strand, the RNA ends up containing an A instead of a G at the 5024 spot. This likely creates issues with folding and/or aminoacylation, and leads to reduced steady state levels of the tRNA. In both the mouse variant and the corresponding human variant, these effects show up most acutely in heart tissue as a classic mitochondrial cardiomyopathy.
Each of the two ZFN proteins were systemically delivered to the mouse via a cardiotropic version of the adenovirus. This particular vector (AAV9.45 serotype16) has a unique coat protein structure that preferentially targets it to the heart over other organs like the liver. The resultant change in mtDNA heteroplasmy was found to be specific to the heart, and sensitive to the precise dose, with 5×1012 viral genomes per monomer per mouse being a typical delivery.
The Moraes lab in Miami is working on a similar strategy to edit mitochondria. They are using different kinds of nucleases called transcription activator-like effector nucleases (Mito-TALENs) which can be made to bind almost any DNA sequence. Reported back-to-back in Nature Medicine, the Moraes results closely corroborate those obtained by the Cambridge group. Like ZFNs, the Mito-TALENs used here aren't necessarily driving the heteroplasmy all the way to zero. In instances and regions where there are high levels of mutant heteroplasmy, sudden depletion of all the local mtDNA would likely be catastrophic.
While finessing heteroplasmy ratios has lots of immediately useful applications, it is important to realize that it won't be a total cure-all if all of a patient's mtDNA happens to be bad. It such a case, there would be no good mtDNA to clonally expand and fill the gaps. Fortunately, other promising techniques like mitochondrial transplantation may eventually be available to seed new healthy mitochondrial populations. One might liken this kind of combo procedure to a bone marrow transplant wherein native populations of white blood cells and their precursors are wiped out and replaced using more desirable donor stock.
As we have recently seen in the highly sensationalized English cases of Charlie Gard at Great Ormond Street Hospital and Alfie Evans at Alder Hey Hospital, the most severe forms of presumptive mitochondrial disease often encompass larger issues that involve the nuclear encoded mitogenome. In Gard's case, the problem was a double mutant dose of a critical subunit for ribonucleotide reductase (RRM2B), which left him unable to build deoxynucleotides for his mitochondria. He needed nucleotides (or nucleoside precursors), or at least a steady supply of healthy mitochondria, but unfortunately couldn't get any real treatment. The Charlie Gard Foundation is currently working to pass Charlie's Law to protect parental rights in these kinds of cases by restricting court involvement when parents seek to bring their kids to another hospital for care.
The case of Alfie Evans was a bit more complex. Despite massive public appeal to find out what was going on, including bold entreaties from Pope Francis and President Trump, no one could figure out what was wrong with the child. It was widely reported that he had a couple of suspicious variants in his mtDNA amidst an underlying mitochondrial disorder. While the particulars here are not yet publicly disclosed, the family did share this particular data and some history with me, and it is far from conclusive, at least to my layperson's amateur eye. However, as we'll see in a minute, there a few things we can learn here.
Martin Picard and mitochondrial founding father Doug Wallace just published a study showing how somatic mtDNA mutations augment the effects of unfavorable mtDNA-nDNA interactions in the progression of cardiomyopathy. Picard also just sent me another recent paper in which he and researchers Amy Vincent and Robert Taylor, among others, show that depletion of mitochondrial DNA spreads across restricted mitochondrial populations in muscle at the subcellular level. In these cells, the subsarcolemmal mitochondria are located at the periphery of the muscle fiber adjacent to the sarcolemma membrane, and a subset of these are distinctly perinuclear. A completely different population, the intermyofibrillar mitochondria, are located between the myofibrils at the Z-band.
The mtDNA-nDNA interactions in the cardiomyopathy study above are precisely the kinds of interactions that may have proved to be critical in Alfie Evan's case. Incidentally, not too long ago, we published a systematic procedure to analyze these kinds of interactions. Using my own mtDNA as an example, and various bioinformatics software tools, I demonstrated that it is now practical to directly generate lines of inquiry to evaluate the larger mito-nuclear genome.
For example, shortly after that was published, a member of the Wallace bioinformatics group wrote me a nice python program that can take a whole genome variant file (.vcf file) and extract just those variants for the genes that appear in the MitoCarta database of nuclear-encoded mitochondrial proteins. This drastically reduces the amount of data that needs to be considered and focuses the scope of the possible. One thing still required for convenience is to extract only the variants that appear in the coding exons. The program and files for that can be found here.
In cases like Alfie's it is difficult to verify facts reported in the media, especially from the outside. Reports indicated that Alfie had been receiving the drug vigabatrin, and at some point thereabouts, he had really started to go south. Vigabatrin is sometimes used for certain types of seizures and inhibits the enzyme that degrades Gaba. Taking that as one possible clue, I wrote a fairly speculative piece ostensibly about Gaba, but particularly about the Gaba transaminase enzyme (ABAT, or Gaba T), nucleotides, and vigabatrin. Without mentioning any names, it was published shortly after Alfie died. Excerpt:
"While it is a close structural analog of GABA, vigabatrin acts as a potent suicide inhibitor of ABAT, but it won't bind all to GABA receptors. GABA levels, mitochondrial levels, and mtDNA levels have all been proven to be very sensitive to precise amounts of vigabatrin given. Undesirable effects of vigabatrin might be expected if given to individuals with pre-existing mitochondrial disease alone."
I am not a doctor, and have no idea what treatments were or were not administered, but I would suppose that giving an inhibitor of Gaba breakdown to someone with a pre-existing deficit in the ability to break down Gaba might drive Gaba levels very, very high. For example, CSF levels can be measured as high as 60 times the normal level in Gaba T variant cases.
In an interesting turn of events, Alfie's parents, Tom and Kate, recently appeared on national TV to announce they are having a second son. After extensive testing, they discovered that they were both carriers of a autosomally recessive mutant Gaba T gene. They further revealed that they were told in June that Alfie had the extremely rare brain condition GABA-transaminase deficiency. In other words, in zeroing in on Gaba T back in May, I was either very lucky, or very right.
The publicly released court documents for Alfie's hearing emphatically said that no more testing was needed, and that they would only have been futile. Hindsight may be 20/20, but in light of the above, it appears that proper early testing, as called for repeatedly and extensively by some individuals, could likely have shown Alfie was doubly recessive for Gaba T.
No one is asserting that mitochondrial genetic engineering is ready to be mainstreamed tomorrow, nor that it would be the whole answer to severe cases of mitochondrial disease. The main point is that in the future, successfully treating kids like Charlie or Alfie will require an integrative strategy that comprehensively considers unique subpopulations of mitochondria throughtout the body. Of particular note, a paper recently posted to Arxiv highlights these issues in more detail. The authors carefully draw a distinction between 'microheteroplasmy,' or intra-cellular mtDNA variance, and 'macroheteroplasmy,' or inter-cell variation. We might additionally introduce the term 'nanoheteroplasmy' for describing situations in which mtDNA varies within a single mitochondrion, or within a single nucleoid of a mitochondrion.
The relevance and possible relation of Alfie's particular mtDNA variants to his particular Gaba T variants in his nuclear mitogenome can be debated. As above, we know many tRNA variants have been linked to conditions like cardiomyopathy or selective damage to the nervous system, and also that many of the variants have yet to be be fully explored.
More information: Payam A. Gammage et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo, Nature Medicine (2018). DOI: 10.1038/s41591-018-0165-9
Sandra R. Bacman et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation, Nature Medicine (2018). DOI: 10.1038/s41591-018-0166-8
© 2018 Medical Xpress