May 16, 2018 report
GABA, GABA, GABA, what does it actually do in the brain?

Gamma-Aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the brain. It is the control knob of all control knobs. But why GABA? What, if anything, might be so special about the molecule?
To look at it, there is nothing inherently peculiar about the small four-carbon backbone structure of GABA. Solvation effects in solution allow GABA to take on five different possible conformations, some compact, and others more extended. At the receptor level, this flexibility means that GABA is a highly 'druggable' target. In other words, pharmaceutical analogs of GABA are more rigidly pocketable in select receptor subsets, and therefore potentially very specific.
If GABA itself is completely ordinary, does it, perhaps by chance, sit in some prized location within metabolism? Some of the apex positions in metabolic cycles are occupied by molecules like Acetyl-CoA and ATP. Acetyl-CoA sits right at the intersection of glycolysis, the TCA cycle, ketone production, β-oxidation of lipids, and fatty acid elongation. It even plugs directly into synthesis of the neurotransmitter acetylcholine. Curiously, there are no major receptor systems associated with this complex molecule that might otherwise keynote its position inside the cell.
ATP has a more condensed footprint than Acetyl-CoA, but it is certainly no slouch. It directly links oxidative phosphorylation with nucleotide metabolism, and is piloted by its own special set of membrane receptors. Like the GABA ecosystem, ATP comes with the full-service suite containing all the expected ionotropic and metabotropic receptor functions. In the case of GABA, these receptor effects have traditionally been neatly grouped into GABAA receptors, which act as channels for chloride ions, and GABAB receptors, which signal through G-protein bound to their undercarriage.
This tidy picture was recently shaken up with reports of GABA directly gating potassium (K) channels. K channels respond to voltage and normally act to hyperpolarize a neuron, or repolarize a it after a spike. The authors of a paper just published in Nature found that the KCNQ receptor family has an evolutionarily conserved spot set aside just for GABA. When GABA is present there, the voltage set point of these channels is shifted to a more polarized state.
KCNQ channels carry the so-called 'M' current, which can be triggered by the agonist muscarine. Because of this receptor-like action, lack of inactivation, and voltage range of operation, these M channels are said to be regulators of neuronal excitability. Often, this just means they convert a neuron from a phasic to a tonic firing pattern. In some cases, neurons with strong M currents might be idealized as voltage controlled oscillators (VCOs). Such a device takes an analog voltage input and generates a variable frequency output. A variation of the VCO, pulse-code modulation, is now the general method of encoding used for uncompressed audio.
While this newfound ability of K ion channel sensitivity is undoubtedly big news in the electrophysiology world, I am going to go out a limb and suggest that the secret of GABA is probably not doing digital audio in your brain. In this void, I think we must dig deeper into what those other kinds of receptors, the slow and roundabout GABAB metabotropic receptors, might be doing. For that matter, we need to break out of the GABA bubble and ask what any G-protein coupled receptor is really doing for the cell.
One clue is that all of these receptors invariably provide for some external control of GTP nucleotide hydrolysis. In that capacity, they are also acting as 'honest signalers' of GTP and GDP status. Furthermore, depending on whether the G protein alpha subunit is itself excitatory or inhibitory, the next second messenger down the line (in this case, adenylate or guanylate cyclase), also deals in nucleotides. Namely, they each cleave a double phosphate from their respective namesake trinucleotide, and then lock the remaining molecule up in a knot until phosphodiesterases eventually cleave it.
Who in the cell would be interested in nucleotides? Neurons are generally stuck in the senescence G0 phase of their cell cycle and aren't worried about having enough nucleotide for the next cell division. There is none coming. Their nuclei only need to maintain a small but steady deoxynucleotide reserve to meet the requirements of DNA repair. In humans, this amount to about 10,000 oxidative lesions per nucleus per day. Therefore, neurons are at liberty to downregulate de-novo nucleotide synthesis enzymes, as well as some of their high-end replication-linked repair pathways.
Mitochondria, on the other hand, are extremely interested in nucleotides. While they have various nucleotide salvage and repair pathways on hand, demand is high—mitochondrial DNA is constantly subjected to mutations and partial deletions, it must be continually refreshed as mitochondria replicate themselves and their nucleoids.
Is there any evidence that GABA signaling and metabolism are directly involved with nucleotide status?
The 'GABA shunt' is a detour on the TCA cycle that figures prominently in the nervous system. The cortex in particular is literally one big fat GABA shunt. The detailed molecular structure of the cortex is what you get when you blow up this particular region of metabolism and build an entire higher brain lobe around it. What I mean by this is that our most refined neural wetware runs a relatively simple GABA-glutamate-glutamine metabolic cycle on an elaborate tripartite synaptic hardware; it is a co-op where three different kinds of mitochondria, those optimized by interneuron, pyramidal cell, and astrocyte, exist in mutual communication with each other across a continually fluctuating membranous border.
The take-off point for the GABA shunt lies at alpha-ketoglutarate in the TCA cycle, and the re-entry point connects up at the succinate. A major branch point, succinate is another apex molecule with a hand in everything. Like acetyl-CoA, it links to all major energy pathways, and even contributes to special orders like cholesterol and heme. Similarly, succinate also comes in an acetylated version, and it sits right between succinate and alpha-KG on the TCA cycle.

The key observation here is that the enzyme that converts it into succinate, succinyl-CoA ligase (aka. succinyl-CoA synthase), has one very important feature: When it is operating in the forward direction of the cycle, it also generates purine nucleotides as part of the deal. Different tissues in the body build slightly different versions of this enzyme by substituting in alternate secondary subunits. The SUCLG2 version makes GTP in anabolic tissue like the liver and kidneys, while and the SULCA2 version makes ATP for catabolic tissue like brain and muscle. When either of these subunits are mutated, you end with mitochondrial disorder. The exact symptoms depend on which variants you have, and in which tissues.
It is worth mentioning here that some of the most puzzling mental phenomena in all annals of medicine occur when there are genetic defects in the enzymes needed in nucleotide metabolism, particularly in those that localize to mitochondria. Cases in point are the bizarre symptomatology of purine-salvage pathways linked to Lesch-Nyhan syndrome, or to stiff-person syndrome, as well as several unique forms of seizure.
On the far side of succinate on the TCA is fumarate. Fumarate can also enter the cycle by way of a reserve purine cycle that kicks into gear in active tissue when metabolic demand is high. Normal operation of the TCA cycle generates fumarate from succinate via succinate dehydrogenase in the inner mitochondrial membrane. This enzyme simultaneously functions as in respiratory chain electron transport as Complex II.

We can see from the diagram above that the GABA shunt appears to bypass this critical nucleotide-generating step of succinyl-CoA synthase. However, researchers have previously found that a GABA transaminase (ABAT) shunt enzyme operates in a complex with the SulcA2 subunit and may also act in the final steps of purine salvage. They also believe that a nucleoside diphosphate kinase (NME4) that posses a mitochondrial localization sequence is part of the complex, although there is still some question as to actual levels of this enzyme in the brain.
Some additional insight into what exactly might be going on was recently provided by the discovery of a new mechanism for generating GABA in the brain without the GABA shunt. Normally, only neurons make GABA from glutamate because only they have the GAD enzyme to do it. But glia can also generate GABA by degrading putrescine via monoamine oxidase B (MAOB) in the mitochondrial outer membrane. This is particularly useful in the cerebellum, where glial Bergman cells, which are responsible for generating and maintaining its highly organized architexture, are abundant. Bergmann glia form palisades aligned with the long axis of the cerebellar folium at a ratio of eight glia to one Purkinje cell, and each glia coordinates around 5000 synapses per Purkinje cell.
Cerebellar ataxias and retinal problems are two common issues that regularly arise across the spectrum of mitochondrial disorders. Since MAOB is coded on the X chromosome, males (who get their X from mom) would inherit it as a package deal together with their mtDNA. This is because we all get our mitochondria exclusively from our mothers. Looking at these kinds of co-inherited linkages is an important tool for probing mitochondrial disease. This information is also important to determine what kinds of drugs should be used in treatments.
For example, vigabatrin is a roundly horrific medicine that causes visual field defects in almost half of the kids who are treated with it. Unfortunately, this is often the only drug that works against some kinds of equally horrible seizures. The researchers mentioned above used vigabatrin experimentally to demonstrate the nucleotide synthesis pathways involved with ABAT. While it is a close structural analog of GABA, vigabatrin acts as a potent suicide inhibitor of ABAT, but it won't bind all to GABA receptors. GABA levels, mitochondrial levels, and mtDNA levels are all been proven to be very sensitive to precise amounts of vigabatrin given. Undesirable effects of vigabatrin might be expected if given to individuals with pre-existing mitochondrial disease.
It was also recently found that a molecule called torin 1 can at least partially correct for some of the primary and incidental effects of vigabatrin. The mechanism involves the ability of GABA to activate mTOR (mechanistic target of rapamycin), which, in turn, blocks elimination of spent or superfluous mitochondria through mitophagy. This also means that more mitochondria with damaged mtDNA will persist. Torin, an inhibitor or mTor (similar to rapamycin), may therefore be a useful tool for dealing with some of the negative effects of vigabatrin in active tissue like the eye and brain.
The authors of the torin study observed some seemingly paradoxical effects in different parts of the brain. For example, they lacked a satisfying explanation for why torin 1 normalized mitochondrial accumulation in the hippocampus, but not in the eye, liver, or parietal cortex, and also why hippocampal mitochondria were increased with vigabatrin, but not in the parietal cortex.
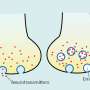
One thing that distinguishes the nervous system from all other non-thinking organs—perhaps its single most defining feature—is its massive, and largely polarized, intracellular conduction of mitochondria. When required, this activity leads to transcellular conduction of mitochondria, often followed by mitophagy, as is well known to be important in the optic nerve.
Such transexudation circuits, perhaps locally between the hippocampus and associated cortical regions, or even more peripherally from the nervous system to other organs, would naturally compensate for metabolic shortfalls that invariably arise in single cells that lack specific enzymes as a result of their genetic differentiation. The GABA shunt is one nice example of this.
Mitochondria are not uniform and faceless like electrons. They age, albeit differentially, sometimes reversibly, and are sorted accordingly in cells and tissues. Spiking in the brain is the direct result of metabolic activity, and seizing the result when things are too far off kilter. We know spikes control mitochondria, their activities, and more visibly, their movements. Now, we also know that one way spikes achieve this control is by manipulating nucleotide metabolism in mitochondria.
More information: Rían W. Manville et al. Direct neurotransmitter activation of voltage-gated potassium channels, Nature Communications (2018). DOI: 10.1038/s41467-018-04266-w
© 2018 Phys.org