Solving a medical mystery: Cause of rare type of dwarfism discovered

For children born with Saul-Wilson syndrome, and their parents, much of their lives are spent searching for answers. First defined in 1990, only 14 cases are known worldwide. And the cause of the syndrome—characterized by short stature, microcephaly (small head), hearing loss and early developmental delays—remained unknown. Today, these individuals have answers.
"This is the news I have been waiting for my whole life," says Monica Zaring, who has Saul-Wilson syndrome. "A diagnosis with Saul-Wilson syndrome is just that—a name. I feel so grateful for all the doctors who never gave up, even when they didn't have answers, and I hope this information will help more people in the future."
Scientists from Sanford Burnham Prebys Medical Discovery Institute (SBP), in collaboration with researchers and clinicians at the National Human Genome Research Institute (NHGRI), the University of Edinburgh, the University of Oregon, and the Nemours/Alfred I. duPont Hospital for Children, have uncovered the cause of Saul-Wilson syndrome: an alteration in the gene that codes for a protein that is part of a component that controls and maintains the cell's protein packager, the Golgi complex (pronounced gol-je). The study published today in the American Journal of Human Genetics.
"Children with Saul-Wilson syndrome and their parents live with many unanswered questions," says Carlos R. Ferreira, M.D., medical geneticist at the NHGRI. "Knowing the underlying cause of the condition is a major step forward for these indviduals and could help scientists find a treatment for the syndrome. This finding also advances our understanding of the genome and Golgi complex's impact on human health, which may help us understand more skeletal disorders."
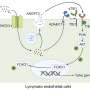
Proteins, the workhorses of our body, are transported inside of our cells via packages called vesicles. These packages travel from one cellular organ, the endoplasmic reticulum (ER), to the Golgi complex, which resembles a stack of pancakes. Keeping the Golgi in working order relies on a protein complex called COG, which has eight subunits.
"The Golgi complex is where proteins 'get ready for the dance,'" says co-first author Zhijie Xia, Ph.D., postdoctoral researcher in the laboratory of Hudson Freeze, Ph.D., co-senior author of the paper and director and professor of the Human Genetics Program at SBP. "Here, proteins are modified in a variety of ways—such as sugars being added or removed—which affects their ultimate function in the body."
Using the latest genetic technology, the scientists analyzed 14 people with Saul-Wilson syndrome. All the individuals had the very same change in just one copy of the gene that codes for COG4 protein, which is part of the Golgi complex. This change in the COG4 gene arose spontaneously (de novo), meaning each parent did not have the mutation. As a result, one amino acid, the building block of proteins, was swapped for another. Specifically, glycine was replaced by arginine.
Further study revealed that in the cells of people with Saul-Wilson syndrome, packages of proteins moved slowly from the ER to the Golgi complex, but then rapidly returned—similar to a delivery truck that drives slowly to your home but speeds back to the warehouse. The size of the Golgi complex was halved. The scientists also found the Golgi complex's ability to add sugars to one protein, decorin, was altered. This protein travels out of the cell and helps support the collagen in skin.
"With the advent of genetic sequencing, we are learning of more conditions that are caused by de novo changes, including autism and epilepsy. Additionally, we are finding more disorders that are caused by heterozygous mutations—which are found only on one gene, versus both genes," says Freeze. "Studying cases of heterozygous, de novo mutations could provide insights into additional mysterious conditions. Our lab is already undertaking a more comprehensive study to understand the impact of the COG4 mutation on protein production and stability."
More information: American Journal of Human Genetics (2018). DOI: 10.1016/j.ajhg.2018.09.003 , www.cell.com/ajhg/fulltext/S0002-9297(18)30314-8