Failing heart cells trigger self-protection mechanism

An unexpected finding that links a structural heart protein to gene regulation following heart stress suggests potential new avenues for developing heart failure therapies.
The work led by University of Iowa heart researcher Long-Sheng Song, MD, focuses on a protein called junctophilin-2 (JP2). Previous work from Song's lab has shown that JP2 is a structural protein that is essential for heartbeat, and that loss or disruption of JP2 is associated with heart failure.
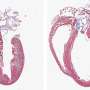
The new study conducted in mice and published online by the journal Science on Thursday, Nov. 8, reveals that under stress conditions, JP2 is cleaved into two fragments. This breakdown of JP2 damages the structural architecture of the cells and disrupts heart cell function. The new study's surprising finding is that one of the newly created fragments of JP2 protects the heart from damage by traveling to the heart cells' nuclei and turning off the expression of genes that promote heart failure.
"We have long known that this protein is a structural protein, important for muscle function, but we never expected it to also have this ability to regulate gene expression," says Song, professor of internal medicine at the University of Iowa Carver College of Medicine. "These findings reveal a previously unknown self-protective mechanism that heart muscle cells possess to counter the damaging effects following cardiac stress."
Heart disease conditions, like high blood pressure, blocked arteries, or heart attack, all put stress on the heart. At the cellular level, this type of stress activates an enzyme that cleaves JP2 into two fragments. The new study shows that the N-terminal JP2 fragment migrates to heart cell nuclei and initiates genetic changes that protect against heart failure. The DNA sequences that allow the fragment to travel to the nucleus and that regulate gene expression are highly conserved among many species from mice to humans.
To prove the beneficial effect of the JP2 fragment, the researchers engineered mice with increased levels of the JP2 N-terminal fragment. These animals were protected from developing heart failure in response to cardiac stress. Conversely, mice genetically engineered to lose the function of the JP2 fragment in the nuclei developed heart failure at an accelerated rate following cardiac stress.
"Our findings suggest that increasing the level of JP2 fragment or functional peptide in the heart might hold promise as a strategy for treating heart failure," says Song, who also is a member of the Fraternal Order of Eagles Diabetes Research Center at the UI and holds a staff appointment with the Iowa City Veterans Affairs Medical Center. "We have secured a patent for the use of the protein fragment, and intend to investigate gene therapy approaches for delivering it to heart cells in preclinical (animal) models of heart failure."
In addition to its role in heart muscle, JP2 is also important and abundant in other types of muscle (skeletal and smooth). The new findings suggest that JP2 fragmentation may play a role to protect against the adverse effects of stress in all types of muscle.
More information: "E-C coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator" Science (2018). science.sciencemag.org/cgi/doi … 1126/science.aan3303