Deletion in mouse neutrophils offers clues to pathogenesis in multiple sclerosis

Multiple sclerosis is an autoimmune disease that damages the insulating sheaths of nerve cells of the central nervous system. People with the disease can lose vision, suffer weak limbs, show degenerative symptoms and exhibit impaired cognition.
While multiple sclerosis has 17 approved therapies to modify the disease, none is able to halt disease progression. Thus, researchers use a mouse model called experimental autoimmune encephalomyelitis, or EAE, to discover disease mechanisms that may translate into treatments for patients with multiple sclerosis. Researchers at the University of Alabama at Birmingham now report in the journal JCI Insight how dysregulated neutrophils cause damage in a severe, mouse model form of EAE called atypical EAE, which attacks cerebellum brain tissue.
"These findings contribute to our understanding of the pathobiology of brain-targeted EAE and document the detrimental role of neutrophils in autoimmune neuroinflammation," said Etty "Tika" Benveniste, Ph.D., and Hongwei Qin, Ph.D., senior authors of the study. Benveniste and Qin are professor and associate professor in the UAB Department of Cell, Developmental and Integrative Biology.
Much evidence from neutrophils points to their detrimental impact in multiple sclerosis. Neutrophils are the most common white blood cells in the body, but their exact function in multiple sclerosis is unclear. Their normal, healthy function is to protect humans, as neutrophils speed to sites of infection or inflammation, aided by their ability to crawl out of the bloodstream and into affected tissues. In everyday life, people encounter them as the most prevalent cells found in pus, as an infection clears.
Several strands of evidence from previous studies at UAB and elsewhere formed the groundwork for this current study. These include 1) UAB researchers and others have shown that brain-targeted, atypical EAE is predominantly a neutrophil-driven disease; 2) dysregulation of a cell-signaling pathway called JAK/STAT is associated with multiple sclerosis and EAE; and 3) a cytokine called granulocyte colony-stimulating factor is known to have a detrimental role in multiple sclerosis, as it correlates with neurological disability and lesion burden in patients.
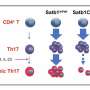
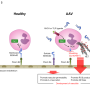
In their experiments, the UAB researchers artificially dysregulated the JAK/STAT signaling system by using mice with a deleted Socs3 gene. Socs3 is a negative regulator of the JAK/STAT pathway; in the absence of Socs3, the JAK/STAT pathway is overly active and promotes inflammation. As a result, mice with Socs3 deletion in their myeloid cells have a severe, brain-targeted, atypical form of EAE that is associated with cerebellar neutrophil infiltration and over-activation of STAT3, one of the seven STAT proteins that function in the JAK/STAT cell signaling pathway.
Using this model, the researchers found that neutrophils from the cerebellum of mice lacking Socs3 showed a hyper-activated phenotype and produced excessive amounts of reactive oxygen species, chemically active compounds that can damage cell structures. However, if mice were given treatments to neutralize the reactive oxygen species, the onset of atypical EAE was delayed and disease severity was reduced.
The mechanisms causing these changes were an enhanced STAT3 activation in Socs3-deficient neutrophils, a hyper-activated phenotype in response to granulocyte colony-stimulating factor, and an increased production of reactive oxygen species after neutrophil priming by granulocyte colony-stimulating factor. Furthermore, when compounds were given to mice to neutralize granulocyte colony-stimulating factor, the incidence and severity of atypical EAE was significantly reduced.
The researchers also sequenced messenger RNA in the Socs3-deficient neutrophils after stimulation by granulocyte colony-stimulating factor to identify the cell-signaling pathways and proteins that were most differentially affected.
"Overall, our work elucidates that hypersensitivity of granulocyte colony-stimulating factor/STAT3 signaling in Socs3-deficient mice leads to atypical EAE by enhanced neutrophil activation and increased oxidative stress, which may explain the detrimental role of granulocyte colony-stimulating factor in multiple sclerosis patients. Furthermore, the work suggests that both granulocyte colony-stimulating factor and neutrophils may be therapeutic targets in MS," Qin and Benveniste said.
The study, "Deficiency of Socs3 leads to brain-targeted EAE via enhanced neutrophil activation and ROS production," is published in the journal JCI Insight.
More information: Zhaoqi Yan et al. Deficiency of Socs3 leads to brain-targeted experimental autoimmune encephalomyelitis via enhanced neutrophil activation and ROS production, JCI Insight (2019). DOI: 10.1172/jci.insight.126520