New intra-nasal imaging to study airways in patients with cystic fibrosis
")
A paper published today in Science Translational Medicine describes a simple, minimally invasive new tool for viewing differences in the nasal airways of cystic fibrosis patients in vivo at a cellular level. The new technique provides high-resolution images of the hair-like structures called cilia that line nasal airways as well as detailed features of the clearance of mucus, which is impaired in people with CF, causing significant morbidity.
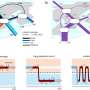
The imaging catheter, which is about 2-millimeters in diameter, uses optical coherence tomography with a resolution of about 1 micrometer—a 50th the size of a human hair—allowing researchers to probe the nasal passages of patients without even sedating them.
"We've done a lot of benchtop imaging with micro-OCT but this is the first time we've been able to use it in people," said co-principal investigator Guillermo Tearney, MD, Ph.D., a pathologist and investigator for the department of Pathology and Wellman Center for Photomedicine at Massachusetts General Hospital (MGH), the Remondi Family MGH Research Institute Chair, and professor of pathology at Harvard Medical School (HMS). "It's unprecedented to see this pathophysiology dynamically in living patients. It will allow us to begin to understand things we never even knew were there."
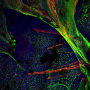
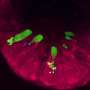
Among the researchers' findings were that in CF patients, the mucus contained more inflammatory cells than in control subjects and it was dehydrated, moving slowly and impacting cilia speed. The researchers were surprised that they were even able to measure the mucus's reflective quality, which they determined was an indicator of viscosity, and that patches of CF patients' cilia and epithelium were ablated. "We thought they were there but just not functioning well," said Tearney, "but in places they were completely gone."
The approximately 30,000 cystic fibrosis patients in the United States have an average life expectancy of about 45 years. The new technique will allow clinicians to earlier diagnose diseases that affect the airways, monitor them and optimize drug therapies for individual patients. "Visualizing abnormal mucus will be a powerful tool," said co-principal investigator Steven M. Rowe, MD, MSPH, director of the Gregory Fleming James Cystic Fibrosis Research Center and a professor of medicine at the University of Alabama at Birmingham. "Now we'll be able to see how various treatments affect the airway—not only agents intended to fix abnormal mucus, which is applicable to many diseases, but also treatments that repair the basic CF defect itself."
 in the mucosa between patients with cystic fibrosis (CF) and healthy controls. Credit: B.R. McDonald et al., Science Translational Medicine (2019)")
The researchers expect micro-OCT to have many applications beyond cystic fibrosis—from diagnosing and treating respiratory ailments like primary ciliary dyskinesia, chronic sinus disease, and chronic obstructive pulmonary disease (COPD) to screening the GI tract for diseases and imaging coronary artery cells. "The goal is to figure out who else it can be used to help," said Rowe, "and make it more broadly accessible as a device."
More information: H.M. Leung el al., "Intranasal micro-optical coherence tomography imaging for cystic fibrosis studies," Science Translational Medicine (2019). stm.sciencemag.org/lookup/doi/ … scitranslmed.aav3505