Team discovers one more piece to the autism puzzle

Mutations in a subunit of a receptor that binds the major inhibitory neurotransmitter GABAA in the brain have been linked, through a common mechanism, to epilepsy, autism and intellectual disability, researchers at Vanderbilt University Medical Center and colleagues report.
The team's discovery, reported last month in the journal Brain, provides more evidence that autism and intellectual disability observed in some patients with severe seizure disorders are not caused by the seizures themselves but arise independently from receptor mutations.
"This work represents a critical piece in the whole puzzle we are working on, the impaired GABAergic pathway in epilepsy, autism and intellectual disability," said the paper's corresponding author, Jing-Qiong "Katty" Kang, MD, Ph.D., associate professor of Neurology and Pharmacology at Vanderbilt University School of Medicine.
Kang said she hopes these insights will help advance development of new, mechanism-based therapies for epilepsy and autism.
Gamma-Aminobutyric acid type A (GABAA) is an inhibitory neurotransmitter that plays an important role in brain development. Mutations in the gene for the beta3 subunit of the GABAA receptor (GABRB3), through which the neurotransmitter acts, are frequently associated with genetic epilepsy syndromes and neurodevelopmental disorders.
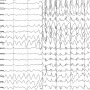
Mutations in the same gene have been linked to a wide spectrum of epilepsy phenotypes (characteristics). Some patients only have mild seizures during childhood that can be easily treated while others have severe epilepsy that is often refractory (does not respond) to multiple medications and has a poor outcome.
The molecular mechanisms responsible for different forms of epilepsy are not known. It's also not clear why mutations in the same gene can produce such widely varying phenotypes.
Working with multiple groups around the world, Kang and her Vanderbilt colleagues have compared the pathophysiology of multiple mutations in different subunits in GABAA receptors.
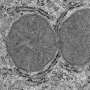
The group previously developed a model of severe genetic epileptic encephalopathy, based on a mutation in another GABAA receptor subunit, GABRG2, and showed that it also causes widespread, age-dependent neurodegeneration.
Recently the researchers identified a mutation in a GABA transporter protein, GAT-1, that causes a similar phenotype to what is observed in patients with GABRB3 mutations.
In this paper, they used high-throughput screening to compare multiple mutations and then focused on two novel mutations in GABRB3 that are associated with different epilepsy severities.
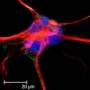
They tested differences of the mutations in non-neuronal cells and rat cortical neurons and in a mouse model with GABRB3 deficiency and identified a common mechanism associated with impairment of the beta3 subunits that occurred in different epilepsy syndromes.
Kang said her group's work contributes to understanding the pathophysiology of genetic epilepsy due to an impaired GABAergic pathway and why mutations give rise to different epilepsy syndromes.
While future studies are needed to clarify the impact of these mutations on brain development, the researchers concluded that impaired activation of the GABAA receptor may contribute to abnormal development of the synapses, the fine functional units between nerve cells that transmit chemical or electrical signals.
"Impaired GABAA receptor function caused by mutations … thus (could) result in complex neurodevelopmental disorders including epilepsy, autism and intellectual disability," they wrote.
The next phase of the research will address how to design treatment to rescue/reverse disease symptoms at an early age and improve outcomes.
More information: Yi-Wu Shi et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes, Brain (2019). DOI: 10.1093/brain/awz250