Understanding gut microbiota, one cell at a time

A population of microorganisms living in our intestine, known as the gut microbiota, plays a crucial role in controlling our metabolism and reducing the risk of conditions such as obesity and diabetes.
Studies have shown that a way to promote the growth of such beneficial microorganisms and modulate their composition for a healthy balance is to add certain forms of fiber, such as inulin, to our diet. However, out of all the tens of trillions of microorganisms in the gut microbiota, it has been difficult to determine which and how microorganisms respond to dietary fiber. This is because current techniques rely on the availability of reference genomes in DNA sequence databases for precise taxonomic classification and accurate functional assignments of specific organisms, but in actuality, an estimated half of the human gut species lack a reference genome. In addition, existing techniques require hours or even days to complete the task.
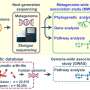
To address this problem, Waseda University scientists devised a novel technique called the single-cell amplified genomes in gel beads sequencing (SAG-gel) platform, which can provide multiple draft genomes of the gut microbiota at once and identify bacteria that respond to dietary fiber at the species level without a need for existing reference genomes. What's more, the advantage of this technique is that it only takes 10 minutes to obtain draft genomes from raw data of whole-genome sequencing since each data is purely derived from individual microbes. This dramatically speeds up the time needed for the process.
"Our new, single-cell genome sequencing technique can obtain each bacterial genome separately and characterize uncultured bacteria with specific functions in the microbiota, and this can help us estimate metabolic lineages involved in the bacterial fermentation of fiber and metabolic outcomes in the intestine based on the fibers ingested," says Masahito Hosokawa, an assistant professor at Waseda University's Faculty of Science and Engineering and corresponding author of this study. "It introduces an enhanced and efficient functional analysis of uncultured bacteria in the intestine."
What the scientists did was to feed mice an inulin-based diet for two weeks and used the technique to randomly capture individual bacterial cells found in the mice's fecal samples into tiny gel beads. The bacterial cells were then individually processed in the gel beads floating in a test tube, and more than 300 single-cell amplified genomes (SAGs), or genomes from a single-cell organism such as bacteria, were obtained by massively parallel sequencing. Because each SAG is composed of tens of thousands of reads on average, it enables extremely cost-efficient whole-genome sequencing of target cells. After quality control and classifying the SAGs, the scientists determined which bacteria were responsible for breaking down inulin and extracting energy from it.
"According to our results, the inulin-rich diet increased activities by the Bacteroides species inside the mouse intestine," Hosokawa explains. "Also, from the draft genomes of newly found Bacteroides species, we discovered the specific gene cluster for breaking down inulin and the specific metabolic pathway for production of specific short-chain fatty acids, a metabolite which is produced by gut microbiota. Findings like these will help scientists in the future to predict the metabolic fermentation of dietary fibers based on the presence and ability of the specific responders."
This technique could be applied to bacteria living anywhere, whether it is inside the human gut, in the ocean, or even in soil. Though there is a need to improve its accuracy since reading the DNA sequence for some gene regions is deemed difficult, Hosokawa hopes this technique will be applied in medicine and industry and be exploited to improve human and animal health.
More information: Rieka Chijiiwa et al, Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota, (2019). DOI: 10.1101/784801