Statisticians work on different fronts to help understand and find solutions in the battle against COVID-19

Statistics is often thought of as an abstract branch of mathematics that relies on complex theoretical models and equations; for the layperson, the applications of such abstract modeling is not always immediately apparent.
But since the COVID-19 outbreak, policymakers and the public have routinely turned to statisticians for insights, from genomic sequencing and clinical trials to testing and contact tracing.
In the Stanford School of Humanities and Sciences, statistics faculty are working on different fronts in the fight against COVID-19. Their work, often in collaboration with Stanford Medicine, could make significant contributions to curbing the pandemic.
This series presents three studies in the Department of Statistics addressing the coronavirus outbreak using different approaches: tracking genetic mutations in the coronavirus, developing a more practical approach to predictive modeling, and designing clinical trials of promising new treatments.
Monitoring mutations
Testing individuals for the coronavirus has been a major challenge. Now biostatisticians are tracking genetic mutations in the virus to estimate the size and spread of COVID-19, and the Centers for Disease Control and Prevention recently announced a major effort to sequence the coronavirus genome.
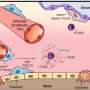
The coronavirus that causes COVID-19 is essentially a single strand of RNA surrounded by a thin membrane of oily lipids and proteins.
RNA, like its close relative DNA, carries the virus's genetic code in a sequence of molecules abbreviated by the letters A, C, G and U. In the coronavirus, the RNA sequence contains about 30,000 letters.
When a person is infected, the invading RNA tricks healthy human cells into cranking out millions of copies of the virus. As the disease spreads and the virus continues to replicate, tiny mistakes, called mutations, occur in the sequence.
For example, the letters A-C-G could be mistakenly copied as A-C-U. These mutated sequences show up in blood samples from people infected with COVID-19, providing scientists important genetic clues about the origin of the pandemic.
"If you have viral samples from different infected individuals, you can estimate how close their viral sequences are genetically by comparing the patterns of shared mutations," said Julia Palacios, an assistant professor of statistics and of biomedical data science. "You can track back in time when pairs of sequences shared a common ancestor."
Hundreds of RNA sequences from COVID-19 viral samples are deposited daily in an open-access data bank, allowing Palacios and her research team to track coronavirus mutations across the globe. These mutated sequences also help scientists determine the size of an infected population and whether it's growing.
"We're looking at the genetic diversity of the virus over time," Palacios explained. "Viruses need to infect individuals to evolve. If the virus infects a large number of individuals, you will observe a large number of mutations. But if the infected population is small, the genetic diversity will remain constant. As you get more sequences, you can see if the trend is still growing, remains unchanged or is starting to decay, in which case the infection may have peaked."
Combining solid genetic data with epidemiological models could lead to more accurate predictions of how the virus will spread in coming months, she added.
"Having a better estimate of the number of cases is very important for epidemiologists to predict what's going to happen in the fall," she said. "But there are so many unanswered questions. For example, our analyses show a highly variable mutation rate suggesting highly heterogeneous strains that evolve at different speeds, but the more data we get, the more questions we can ask and try to answer."
For Palacios, understanding how the virus evolves is more than an academic exercise.
"COVID-19 is affecting everybody, and we're trying to find ways to help," she said. "That's the motivation for my group to work on this."