Researchers find potential treatment for Rett Syndrome

An experimental cancer drug can extend the life of mice with Rett Syndrome, a devastating genetic disorder that afflicts about one of every 10,000 to 15,000 girls within 6 to 18 months after birth, Yale researchers report June 10 in the journal Molecular Cell.
In addition, the drug JQ1 also restores the cellular function of neurons in human models of the disease. Rett Syndrome causes severe deficits in language, learning and other brain functions and eventually leads to death, often during teenage years.
The Yale team—led by senior author In-Hyun Park, associate professor of genetics, and a researcher at Yale's Child Study Center and Stem Cell Center—wanted to know how a mutation in gene MECP-2 causes the severe disruption to neuronal functions in the cortex of Rett Syndrome patients.
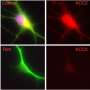
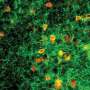
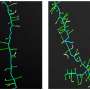
They created a human brain organoid containing this mutation from embryonic stem cells and found severe abnormalities in multiple brain cells. A type of brain cell called interneurons, which regulate the brain's excitatory neurons, was particularly impacted by the mutation.
The lab then screened a variety of compounds and found that one drug, JQ1, corrected abnormalities found in interneurons of the Rett Syndrome model. The drug has been investigated in several experimental trials as a potential cancer treatment.They then tested the drug in mice models of Rett Syndrome and found that the treated mice lived about twice as long as those not receiving the drug.
Park said the research paves the way for additional research on potential new therapies for Rett Syndrome, for which there are currently no effective treatments.