Investigational new therapy prevents onset of Dravet syndrome symptoms in mice

In a development that may finally offer hope to children with Dravet syndrome and their parents, a promising investigational new therapeutic appears to alter the destructive course of the deadly disease in a mouse model.
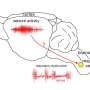
Lori Isom, Ph.D., Chair of the U-M Department of Pharmacology and Professor of Molecular & Integrative Physiology, and Neurology and her team have spent several years tracing the developmental pathway of Dravet syndrome, a debilitating genetic disease that causes intractable seizures and can lead to sudden death. Dravet syndrome variants lead to a decrease in the level of a crucial protein that normal brain cells use to control the flow of sodium ions into cells. Neurons use these sodium channels to transmit signals back and forth.
Patients with Dravet syndrome have variants in a sodium channel gene, SCN1A, that in general result in there being only half the amount of the corresponding functional protein, Nav1.1, expressed in the brain. "With only 50 percent SCN1A expression in the brain, you have a severe epileptic disease with a high risk of sudden death," Isom says.
The team's goal was to increase the gene's expression back up to 100 percent in neurons using an antisense oligonucleotide (ASO) that was developed by Stoke Therapeutics, a biotechnology company. In collaboration with Stoke, Dr. Isom and her team tested the ASO called STK-001 in a Dravet syndrome mouse model.
"The brain normally uses sections of genes called poison exons to dial up or down the expression of a particular gene, as an internal regulatory mechanism," explains Isom. "We are taking advantage of a poison exon in SCN1A to increase its expression." Unlike using CRISPR, viruses, or other forms of gene editing, ASOs are reversible. "The problem with gene therapy is you are changing something forever, which is risky especially with the brain. If you make a mistake, there's no going back."
Injecting the ASO into the brains of mice that carry only one healthy copy of the SCN1A gene two days after they were born led to 97 percent survival out to 90 days, while just 23 percent of the untreated mice survived during the same time range. "Our thought is that early intervention with an ASO in mice may be able to change brain excitability," says Isom, "And if that's also true for kids with Dravet...that would be incredible." The results are published in the journal Science Translational Medicine.
Based on the encouraging findings of the mouse study, Stoke has recently launched a clinical study to begin evaluating STK-001 in children and adolescents with Dravet syndrome.
Isom notes that the mouse study could have even wider implications for treating the genetic cause of generalized epilepsy, especially for the third of patients whose epilepsy is not controlled with traditional drugs. "We just need to start over and think about epilepsy in a new light—to get at the genetic source, rather than being reactive and trying to treat something that has already happened."
More information: "Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome" Science Translational Medicine (2020). stm.sciencemag.org/lookup/doi/ … scitranslmed.aaz6100