How night vision is maintained during retinal degenerative disease

New insight on how people with retinal degenerative disease can maintain their night vision for a relatively long period of time has been published today in the open-access eLife journal.
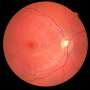
The study in mice suggests that second-order neurons in the retina, which relay visual signals to the retinal ganglion cells that project into the brain, maintain their activity in response to photoreceptor degeneration to resist visual decline—a process known as homeostatic plasticity. Rod photoreceptors are the cells responsible for the most sensitive aspects of our vision, allowing us to see at night, but can be lost during retinal degenerative disease.
The new findings pave the way for further research to understand how our eyes and other sensory systems respond and adapt to potentially compromising changes throughout life.
"Neuronal plasticity of the inner retina has previously been seen to occur in response to photoreceptor degeneration, but this process has been mostly considered maladaptive rather than homeostatic in nature," explains co-first author Henri Leinonen, a postdoctoral researcher at the University of California, Irvine, US. "Our study was conducted at a relatively early stage of disease progression, while most previous studies focused on severe disease stages, which may account for the discrepancy. Very recently, several studies using triggered photoreceptor loss models have shown adaptive responses in bipolar cells—cells that connect the outer and inner retina. But whether such adaptation occurs during progressive photoreceptor degenerative disease, and whether it helps to maintain visual behavior, was unknown."
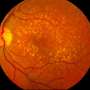
To address this question, Leinonen and colleagues studied a mouse model of retinitis pigmentosa. This is the name given to a group of related genetic disorders caused by the P23H mutation in rhodopsin, a protein that enables us to see in low-light conditions. Retinitis pigmentosa causes the breakdown and loss of rod-shaped photoreceptor cells in the retina, leading to difficulties seeing at night.
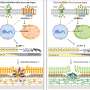
The team combined whole-retinal RNA-sequencing, electrophysiology and behavioral experiments in both healthy mice and those with retinitis pigmentosa as the disease progressed. Their experiments showed that the degeneration of rod photoreceptors triggers genomic changes that involve robust compensatory molecular changes in the retina and increases in electrical signaling between rod photoreceptors and rod bipolar cells. These changes were associated with well-maintained behavioral night vision despite the loss of over half of the rod photoreceptor cells in mice with retinitis pigmentosa.
"This mechanism may explain why patients with inherited retinal diseases can maintain their normal vision until the disease reaches a relatively advanced state," says co-first author Nguyen Pham, Graduate Research Assistant at the John A. Moran Eye Center, University of Utah Health, Salt Lake City, US. "It could also inspire novel treatment strategies for diseases that lead to blindness."
"Our results suggest retinal adaptation as the driver of persistent visual function during photoreceptor degenerative disease," concludes senior author Frans Vinberg, Ph.D., Assistant Professor at the John A. Moran Eye Center, University of Utah Health. "Additional research is now needed to discover the exact homeostatic plasticity mechanisms that promote cellular signaling and visual function. This could help inform the development of potential new interventions to enhance homeostatic plasticity when needed."
More information: Henri Leinonen et al, Homeostatic plasticity in the retina is associated with maintenance of night vision during retinal degenerative disease, eLife (2020). DOI: 10.7554/eLife.59422