February 4, 2021 feature
Drugging the undruggable, improbable new targets for lung cancer therapy
. DOI: 10.1016/j.xcrm.2020.100186")
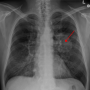
The growth of solid tumors is frequently driven by mutations in key proto-oncogenes. For non-small-cell lung cancers (NSCLC), somatic mutations in the KRAS (Kirsten RAt Sarcoma virus) gene turn it into an oncogene that renders tumors resistant to common chemotherapies like erlotinib (Tarceva) or gefitinib (Iressa).
Previously, KRAS was considered to be "undruggable" because the surface of the tiny protein had no deep pockets for drug interaction with potential small molecule inhibitors. Since many NSCLCs rely on a constitutively activated mutant KRAS, researchers have continued to explore KRAS and its downstream signaling pathways as possible targets. That research has finally begun to pay off. Complementary approaches to NSCLC lung cancer that collectively embrace immune self-defenses within the context of the larger KRAS ecosystem have now come fully into view.
Together, they flesh out a therapeutic microcosm of tumor biology that can be copied and modified to serve as a blueprint for treating many cancer types, each sustained by their own unique oncogenic drivers. In an article in Cell Reports Medicine researchers outline four ways to combat KRAS-dependent NSCLC: immune checkpoint inhibitors, KRAS neoantigen targeting, direct KRAS inhibitors and KRAS signaling inhibitors.
Immune checkpoint inhibitors (ICIs) are already a standard of care for many cancers. PD-L1 (programmed cell death ligand 1) expression has been associated with KRAS-specific alterations in KRAS NSCLC cell lines and patient tumor tissue. NSCLC tumors frequently overexpress PD-L1; therefore, anti-PD-1 therapies, like pembrolizumab (Keytruda) can be beneficial either alone or alongside inhibitors of common KRAS variants. For example, Amgen's AMG510 inhibitor (specific against G12C variants) promotes a pro-inflammatory tumor microenvironment that generates long-term T-cell tumor responses when given with ICIs.
KRAS mutations, like G12C, can create new epitopes that can be recognized by the immune system as foreign proteins. Since the immune system was never trained on a version of KRAS that only mutated in the cancer cells later on, they will be seen as neoantigens that should be attacked. The problem is that the immune system generally needs a little help by the time a tumor is detected. The goal of an immunotherapy is to either elicit a direct T cell response to these neoantigens through vaccination, or via an adoptive T cell therapy in which cells are externally educated, expanded and reintroduced. Moderna Therapeutics and Merck already have mRNA cancer vaccines in trials against KRAS G12C, G12D, G13D and G12V. The vaccine is encoded as a single RNA molecule neoantigen concatemer that should be optimally presented on HLA-A11:01 and/or HLA-C∗08:02 receptors to enhance CD8+ T cell response.
We have seen above how direct inhibitors of mutant KRAS can be effective, especially when given with ICIs. The main benefit is that cells with non-mutated, wild-type KRAS should not be affected by the drugs, reducing side effects. Another strategy against KRAS capitalizes on the fact that it is localized to the plasma membrane for function by farnesylation of specific amino acid side chains. Unfortunately, farnesyltransferase inhibitors have so far yielded poor results in clinical trials. Similarly, small-molecule inhibitors designed to prevent GTP-KRAS interactions have also failed, primarily due to the high affinity of GTP for KRAS. Alternatively, pan-KRAS inhibitors, like BBP-454 (BridgeBio Pharma) have now been developed that can bind to previously unrecognized binding pockets. Another pan-KRAS inhibitor, BI 1701963, interferes with KRAS binding to SOS1, a guanine nucleotide exchange factor essential in activating KRAS.
Several genes have already been identified via synthetic lethal screenings that are involved in KRAS signaling mechanisms in NSCLC. These include TANK-binding kinase 1 (TBK1), cyclin-dependent kinase 4 (CDK4), and B-cell lymphoma-extra large (BCL-XL). Targeting their expression with either trametinib (MEK1/2 inhibitor) or navitoclax (BCL-XL/BCL-2 inhibitor) resulted in significant apoptosis and tumor regression in xenograft models. KRAS signaling also induces fatty acid synthesis by activating lipogenesis and ERK2 kinase activity. Inhibition of fatty acid synthesis enzymes with cerulenin in KRAS mutant lung adenocarcinoma cell lines showed a significant decrease in cell growth.
The full response of a NSCLC patient to chemotherapy depends on the presence of variants in other genes besides KRAS. For example, those who harbor an EGFR (epidermal growth factor receptor) mutation have a 60% response rate to erlotinib. It appears that to some extent, mutation of KRAS and EGFR are mutually exclusive. Lung cancer patients who are positive for KRAS mutation, but not EGFR, have poor chemotherapy response. As the full mutational landscape for KRAS is now rapidly coming to light, effective treatment may involve a combination approach of direct KRAS targeting and indirect targeting through synthetic lethality of downstream effectors. Synthetic lethality arises when a combination of deficiencies in the expression of two or more genes leads to cell death, while deficiency in only one of these genes does not. Mutations, epigenetic changes and inhibitors can all create conditions of deficiency.
More information: Ravi Salgia et al. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC), Cell Reports Medicine (2021). DOI: 10.1016/j.xcrm.2020.100186
© 2021 Science X Network