New high-throughput method greatly expands view of how mutations impact cells

There are millions of mutations and other genetic variations in cancer. Understanding which of these mutations is an impactful tumor "driver" compared to an innocuous "passenger," and what each of the drivers does to the cancer cell, however, has been a challenging undertaking. Many studies rely on bespoke, time-consuming, gene-specific approaches that provide one-dimensional views into a given mutation's broader functional impacts. Alternatively, computational predictions can provide functional insights, but those findings must then be confirmed through experiments.
Now, in a report published in Nature Biotechnology, a research team at the Broad Institute of MIT and Harvard has unveiled a massive-scale, high resolution method for functionally assessing large numbers of protein-coding mutations simultaneously, one that returns rich phenotypic information and which could potentially be used to study any mutation in any gene in cancer and perhaps other diseases. Their results, gained through proof-of-concept experiments with cancer cell lines, also show that individual mutations can have a spectrum of effects not only on their impacted genes but also on molecular pathways and cell state as a whole, and add nuance to the long-accepted practice of dividing cancer mutations into so-called "drivers" and "passengers."
"When you look at the genetic data from patients' tumors, you see that the majority of cancer-associated mutations are actually quite rare, which means we have few insights into what these mutations do," said Jesse Boehm of the Broad's Cancer Program, who was co-senior author of the study with Aviv Regev, a Broad core institute member now at Genentech, a member of the Roche Group. "For cancer precision medicine to become a reality, we need a firm understanding of the function of each mutation, but a major challenge has been defining an experimental approach that could be implemented in the lab at the scale required. This new method may be the tool we need."
The new method, called single-cell expression-based variant impact phenotyping (sc-eVIP), builds on Perturb-seq—an approach developed in 2016 by Regev and colleagues for manipulating genes and exploring the consequences of those manipulations using high-throughput single-cell RNA sequencing—and eVIP, a method also developed in 2016 by Boehm and colleagues for profiling cancer variants at low scale using RNA measurements. While Perturb-seq assays originally relied on CRISPR to introduce mutations into cells, the sc-eVIP team adopted an overexpression-based approach, engineering DNA-barcoded gene constructs for each mutation of interest and introducing them into pools of cells in such a way that the cells expressed the mutated genes at higher-than-normal levels.
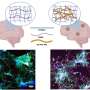
By then recording each perturbed cell's expression profile using single cell RNA sequencing, the team could both identify which mutation a given cell carried (based on the constructs' unique barcodes) and examine the mutation's broader impact on the cell's overall expression state. This approach provides a highly detailed view of a mutation's impact on a variety of molecular pathways and circuits, and does not need to be adapted for each new gene studied.
"In a sense, we're using the cell as a biosensor," said Oana Ursu, a postdoctoral fellow in the Regev lab, formerly within the Broad's Klarman Cell Observatory and now at Genentech, and co-first author of the study with JT Neal, a senior group leader in the Broad's Cancer Program. "By looking at the expression changes that take place when we overexpress a mutated gene, we can learn whether it has a meaningful impact. But also, we can compare and categorize variants based on the changes they trigger, and look for patterns in the biology they affect."
"Most of the technologies developed for interpreting coding variants up to now have been very scalable, but have had relatively simple readouts like cell viability or maybe looked at a single trait. Their information content has been low, and it takes a lot of work to optimize them," said Neal. "With sc-eVIP, we've engineered a comprehensive approach that's high throughput and information-rich, which could be a real boon for large-scale variant-to-function studies."
To test sc-eVIP's potential, the team chose to study TP53—the most commonly mutated gene in cancer—and KRAS—which encodes a key oncogene responsible for abnormal growth of many cancers. Neal, Ursu, and their collaborators generated constructs containing 200 known TP53 and KRAS mutations (including cancer-associated mutations and control mutations known to leave gene function unaffected) and introduced them into 300,000 lung cancer cells, and captured each cell's individual expression profile. Based on those profiles, the team categorized each mutation as either "wildtype-like" (that is, effectively functionally indistinguishable from the unmutated gene) or "putatively impactful," from there further defining mutations based on whether they reduced or enhanced the gene's function.
The profiles also revealed each mutation's broader impact on cell state, based on how the activity of a variety of pathways changed across single cells. For instance, the sc-eVIP data revealed KRAS mutations that fall along a continuum in how they impact cell state at the population level, from having no impact to influencing subtle shifts in cellular abundances to causing outright activation or repression of key pathways in a majority of cells. These findings suggest that different mutations within the same gene can influence cell state along a spectrum of impact.
"The cancer community has long embraced a binary conceptual framework of 'driver' mutations, ones that promote cancer development and progression, versus 'passenger' mutations, which are completely inert and just happened to arise along the way," Boehm noted. "These initial findings suggest that biologically those categories are likely overly simplistic, that there's actually a continuum of functional impact from inert to completely tumorigenic."
While the team focused on cancer-associated genes and mutations for this study, they noted that sc-eVIP is gene-agnostic, highly scalable, and that using single cell RNA sequencing as a readout offers an efficient and generalizable approach to producing rich phenotypic data. They also calculated that it should be possible to thoroughly characterize most mutations with only 20 to a few hundred cells. Based on those numbers, it may be possible with sc-eVIP to generate a first-draft functional map of more than 2 million variants in approximately 200 known cancer genes with 71 million cells.
"If we can map where every cancer-associated variant fits on the continuum of impact in a variety of cancers and cell types," Boehm said, "we'll have a much better grasp of how the interplay of variants affects cell state, which in turn affects cancer development, growth, and response. Such knowledge would represent a true advance toward cancer precision medicine."
More information: Oana Ursu et al, Massively parallel phenotyping of coding variants in cancer with Perturb-seq, Nature Biotechnology (2022). DOI: 10.1038/s41587-021-01160-7