Proportion of mutations in cellular protein drives neurodegeneration

Tubulinopathies are a group of rare developmental and degenerative diseases that primarily affect the nervous system. While research has linked genetic mutations with these diseases, it's less clear how these mutations specifically affect cells and trigger dysfunctions.
In a new study, Yale scientists identify one particular genetic mutation that impairs brain cells. They also reveal the cellular scenario that triggers this response, a phenomenon in which the amount of mutated protein begins to outweigh the number of proteins that are not mutated, leading to excessive cell death. The finding could lead to treatments for tubulinopathies going forward.
The findings were published Feb. 16 in the journal Science Advances.
Tubulinopathies are caused by mutations in the genes that code for tubulins, critical proteins that form the building blocks of microtubules, hollow fibers that essentially serve as the skeletons of cells and enable such critical functions as cell division and the maintenance of cell shape. "They're kind of like the railroad tracks inside cells," Karel Liem Jr., an associate professor of pediatrics at the Yale School of Medicine and senior author of the study, said of microtubules.
And in the nervous system, microtubules form axons, the projections of neurons that conduct signals and allow neurons to communicate with each other.
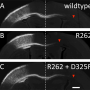
To better understand the connection between tubulin mutations and the emergence of nervous system disease, Liem and his colleagues studied the genetic line of mice that displayed neurological complications similar to those observed in humans with particular neurological disorders. Compared with their healthy counterparts, these mice had smaller cerebellums, less white matter throughout the brain, exhibited problems related to movement, and had frequent seizures.
The researchers discovered two main neurological problems in these mice. They had significantly less myelin, a fatty substance that coats axons and helps signals move through neurons. And their smaller cerebellums, they found, were due to rampant cell death that occurred after development.
"Granule cells died in spectacular fashion," said Liem. "These are the most numerous neurons in the brain and within a couple of weeks, they're all gone in these mice."
Tubulin proteins, those building blocks of microtubules, come together in pairs—an alpha-tubulin links to a beta-tubulin and pairs of alpha- and beta-tubulins string together to form microtubules. There are at least eight types of alpha-tubulins and eight types of beta-tubulins, each encoded by separate genes. When the researchers went looking for what might underlie the neurological defects in the mice, they came across a mutation in a gene coding for one type of beta-tubulin known as tubulin beta-4a.
To better understand the role this tubulin played in the mouse brain, the researchers generated mice that lacked the gene for tubulin beta-4a altogether. Interestingly, these mice did not have any neurological defects; they remained healthy.
"The mice missing tubulin beta-4a were fine," said Liem. "What happens is that there is transcriptional compensation from other tubulin isotype genes and other tubulins get incorporated into microtubules instead. The missing tubulin beta-4a is simply replaced."
But while missing tubulin beta-4a did not cause dysfunction, mutated tubulin beta-4a did. And as the researchers delved further, they found that more mutated tubulin led to more severe neurological defects.
"What we think is going on is that when you have a dominant missense mutation in a tubulin, the tubulin gets incorporated into microtubules and it causes microtubule dysfunction," Liem explained. "Based on our findings, it appears that the severity of dysfunction is due to the expression ratio of the mutated tubulin isotype to all of the tubulins."
In the case of tubulin beta-4a, this means that if a cell in the mouse has a high number of mutated tubulin beta-4a in relation to its pool of available beta-tubulins, then the mouse is going to have more severe disease.
This observation also sheds light on why mutations in tubulin beta-4a seem to have their strongest effects in the nervous system. The brain cells most affected in this study expressed tubulin beta-4a more than they did other types of beta-tubulins. "So if there's a mutation in the gene for tubulin beta-4a, these cells are in trouble because the majority of their beta-tubulins are mutated. Cells that have more of a balance in their pool of beta-tubulins would be less susceptible to a tubulinopathy."
These findings provide a mouse model for studying different tubulinopathies and their cell-specific effects, Liem said. They also help researchers understand the human diseases linked to mutations in tubulin beta-4a, and provide insights into potential therapeutics.
"If we could reduce the expression of the mutated tubulin gene, and do so early, we might be able to stop the neurodegenerative effects of the mutation," said Liem. "Whether it's possible to regenerate what was already lost is another question that has to be answered."
Liem emphasized that this type of therapeutic would be difficult to achieve. "But we're thinking about this," he said.
More information: Sofia Fertuzinhos et al, A dominant tubulin mutation causes cerebellar neurodegeneration in a genetic model of tubulinopathy, Science Advances (2022). DOI: 10.1126/sciadv.abf7262