Data published elucidating a mechanism for the neuroprotective potential of pridopidine

Prilenia Therapeutics B.V., a clinical stage biotechnology company focused on the urgent mission to develop novel therapeutics to slow the progression of neurodegenerative and neurodevelopmental disorders, today announced publication of research in Autophagy which support pridopidine's potential neuroprotective properties by enhancing autophagy in an amyotrophic lateral sclerosis (ALS) model.
Pridopidine is an oral, highly selective and potent Sigma-1 Receptor (S1R) agonist currently in late-stage development for the potential treatment for Huntington's disease (HD) and ALS. In clinical studies to date, pridopidine (45mg twice daily) has exhibited a safety and tolerability profile similar to placebo.
The S1R is highly expressed in the brain and spinal cord where it regulates several processes that are commonly impaired in various neurodegenerative diseases. Activation of the S1R by pridopidine stimulates multiple cellular pathways, including autophagy, which are essential to neuronal function and survival, and may lead to neuroprotective effects.
Autophagy is the process by which a cell eliminates old or damaged proteins. By improving autophagy, pridopidine may reduce the toxic protein aggregation that is a hallmark of neurodegenerative diseases such as HD, ALS, Alzheimer's disease and Parkinson's disease.
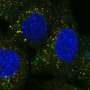
Shuttling of molecules from the cytoplasm to the nucleus (nucleocytoplasmic shuttling), via the nucleopore complex is essential for neuronal function and survival. A critical step in autophagy is the transport of the Transcription Factor EB (TFEB) into the nucleus, where it upregulates expression of autophagy-related genes. This nucleocytoplasmic process is impaired in various neurodegenerative diseases, including ALS.
"This study confirms that pridopidine plays an important role in maintaining the health of a neuronal cell by activating the S1R which in turn interacts with specific proteins to initiate the autophagic process," said Tsung-Ping Su, Ph.D., Chief, Cellular Pathobiology Section, National Institute on Drug Abuse (NIDA) located in Baltimore, MD.
"While this paper focuses primarily on an ALS model, our results also suggest promise for a pharmacological approach targeting S1R across diverse diseases in which autophagy is impaired."
Highlights from the article include:
- This new research shows that pridopidine activates the S1R by facilitating its disassociation from the ER protein BiP, freeing the S1R to interact with other protein partners.
- The activated S1R stabilizes the nucleopore complex by interacting with POM121 (a central nucleopore protein), facilitating the translocation of TFEB into the nucleus.
- TFEB translocation into the nucleus initiates the autophagic process by upregulating the expression of autophagy genes, leading to neuroprotection.
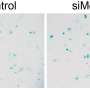
G4C2 repeats within the c9orf72 gene are the most common cause of familial ALS (fALS). This study utilized a cell model of motor neurons over expressing G4C2 repeats that destabilizes the nucleopore and reduces the nucleocytoplasmic transport of TFEB, leading to impaired autophagy and neurodegeneration. In this model, pridopidine activation of the S1R rescues TFEB shuttling and upregulates autophagy, leading to neuroprotection.
"Our collaborations with leading researchers as well as our close working relationships within the HD and ALS communities are important as we enhance our understanding of the mechanism of action of pridopidine and its impact on neurodegenerative conditions," said Dr. Michael R. Hayden, CEO and Founder of Prilenia. "The data published in Autophagy reinforce findings from earlier studies indicating that pridopidine may offer neuroprotective effects. We look forward to sharing additional information about pridopidine as data from our late-stage clinical studies become available."
This new research adds to the growing understanding of pridopidine's mechanism of action as a highly selective S1R agonist, which was previously shown to improve cellular functions impaired in neurodegenerative disease such as mitochondrial function, ER stress, energy production, trophic factor availability and synaptic plasticity.
More information: Shao-Ming Wang et al, Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine, Autophagy (2022). DOI: 10.1080/15548627.2022.2063003