Organoids reveal similarities between myotonic dystrophy type 1 and Rett syndrome

Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy, characterized by progressive muscle wasting and weakness and caused by abnormally repetitive DNA segments that are transcribed into toxic molecules of RNA. Instead of ferrying a gene's instructions for translation into proteins, these RNA molecules accumulate in cells, disrupting cellular machinery.
Rett syndrome (RS) is a rare genetic neurological disorder that affects the way the brain develops, resulting in progressive loss of motor skills and language early in life.
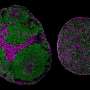
Writing in the June 29, 2022 online issue of Science Translational Medicine, researchers at University of California San Diego School of Medicine used three-dimensional brain organoids—self-organized tissue grown from stem cells that mimics neurological functions—to discover fundamental similarities between DM1 and RS, and perhaps therapeutic opportunities.
"We turned to 3D brain organoids that simulate the developing human cortex to study the effects of the CTG repeat expansion on neuronal processes," said first author Kathryn Morelli, Ph.D., a fellow in the lab of senior author Gene Yeo, Ph.D., professor of cellular and molecular medicine at UC San Diego School of Medicine.
"It's a model that can be made from induced pluripotent stem cell lines from real DM1 patients that carry these toxic RNA aggregates. It mimics cortical development in utero."
Unlike other types of muscular dystrophy, patients with DM1 often exhibit progressive neurocognitive symptoms, with learning and social impediments that can appear similar to autism spectrum disorders. Recent clinical data has shown that the higher the number of inherited DNA repeats, the earlier the onset of symptoms—and the greater the impact of the disease on the central nervous system.
Modern DM1 treatments target only skeletal and heart muscle defects. Research by Yeo and colleagues has shown that RNA-targeting CRISPR/Cas proteins can bind to repetitive RNA in live human cells and reverse markers of disease in the skeletal muscle of mouse models of DM1.
"Still, the absence of a cellular model of the human brain limited our understanding of how toxic RNA can cause cognitive symptoms, and hindered efforts to develop an effective holistic therapy," said Morelli.
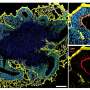
In the latest study, researchers packaged a compact RNA-targeting CRISPR/Cas protein into viral vectors, then added them to DM1 brain organoids. They found that the proteins destroyed toxic RNA aggregates, with scientists able to observe and control the cascade of events.
The team has focused on a model in which toxic RNA traps a special class of proteins called RNA binding proteins, or RBPs. "In cortical organoids, we were surprised to find that another RBP called CELF2 protein was dysregulated in glutamatergic neurons, which are responsible for excitatory signaling in the brain," said Yeo.
Using the enhanced cross-linking and immunoprecipitation technologies pioneered in the lab, Morelli and colleagues discovered that CELF2 did not bind its normal targets: genes in the methyl-CpG binding protein 2 (MECP2) pathway that are crucial for neuron function. Mutations that result in the loss of MECP2's normal function cause RS.
The findings, said the authors, suggestion a possible convergence in neurodevelopment defects in DM1 and RS. Morelli noted that clinical trials are underway to evaluate the therapeutic potential of N-methyl-d-aspartic acid (NMDA) antagonists for treating patients with RS. NMDA receptors are believed to be important in controlling synaptic plasticity and mediating learning and memory functions.
In DM1 organoids, Morelli found that NMDA antagonists reversed key features of the disease, suggesting that targeting NMDA receptors might ameliorate cognitive impairments in young patients with DM1, and substantially improve their quality of life.
Co-authors include Wenhao Jin, Shashank Shathe, Assael A. Madrigal, Krysten L. Jones, Joshua L. Schwartz, Tristan Bridges, Jasmine R. Mueller, Archana Shankar, Isaac A. Chaim, all at UC San Diego; and John W. Day, Stanford University.
More information: Kathryn H. Morelli et al, MECP2-related pathways are dysregulated in a cortical organoid model of myotonic dystrophy, Science Translational Medicine (2022). DOI: 10.1126/scitranslmed.abn2375