Untangling the role of tau in Alzheimer's disease
 h-tau loaded in presynaptic terminals inhibited excitatory synaptic transmission. Credit: Tetsuya Hori et al, eLife (2022). DOI: 10.7554/eLife.73542")
Alzheimer's disease is a brain disorder that causes neurons to die, slowly destroying memory and thinking skills. It's the most common type of dementia, impacting an estimated 50 million people worldwide, and is a particularly serious issue for Japan's super-aged society. Despite its prevalence, the causes remain poorly understood and treatment options are limited.
Now, a team of scientists in Japan has revealed how excess tau—a key protein implicated in Alzheimer's disease—impairs signaling between neurons in the brains of mice. The study, published recently in eLife, could open new pathways for treating the symptoms and even halting the progression of Alzheimer's disease and other neurodegenerative disorders.
Tau is produced in neurons, where it binds to and promotes the assembly of microtubules—long, thin filaments that maintain cell structure and provide pathways for transport within the cell. Tau usually exists in either this bound state, or it is dissolved in the liquid that fills the cell.
However, in some neurological disorders, most famously in Alzheimer's disease, levels of soluble tau in certain brain regions become too high, and it aggregates into insoluble structures called neurofibrillary tangles.
"A lot of scientists focus on the impact of these visible neurofibrillary tangles that are a hallmark of Alzheimer's, but actually, it's the invisible levels of soluble tau that correlate most closely with cognitive decline," said Professor Tomoyuki Takahashi, senior author of the study, and head of the Cellular and Molecular Synaptic Function Unit at OIST.
The research began ten years ago, when his team looked at the effect of high levels of soluble tau on signal transmission at the calyx of Held—the largest synapse in mammalian brains. Synapses are the places where two neurons make contact and communicate. When an electrical signal arrives at the end of a presynaptic neuron, chemical messengers, known as neurotransmitters, are released from membrane "packets" called vesicles into the gap between neurons. When the neurotransmitters reach the postsynaptic neuron, they trigger a new electrical signal.
Using mice, Prof. Takahashi's research team injected soluble tau into the presynaptic terminal at the calyx of Held and found that electrical signals generated in the postsynaptic neuron dramatically decreased.
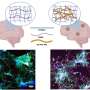
The scientists then fluorescently labeled tau and microtubules and saw that the injected tau caused new assembly of many microtubules in the presynaptic terminal.
However, when they injected a mutant tau protein instead that lacked the binding site necessary to assemble microtubules, there was no effect on synaptic transmission.
"This told us that the decrease in synaptic signaling was clearly linked to these newly assembled microtubules," explained Prof. Takahashi.
A second important clue was that elevated tau only decreased the transmission of high-frequency signals, while low-frequency transmission remained unchanged. High-frequency signals are typically involved in cognition and movement control.
The researchers suspected that such a selective impact on high-frequency transmission might be due to a block on vesicle recycling.
Vesicle recycling is a vital process for the release of neurotransmitters across the synapse since synaptic vesicles must fuse with the presynaptic terminal membrane, in a process called exocytosis. These vesicles are then reformed by endocytosis and refilled with neurotransmitter to be reused. If any of the steps in vesicle recycling are blocked, it quickly weakens high-frequency signals, which require the exocytosis of many vesicles.
The scientists found that high levels of soluble tau primarily impaired endocytosis. The lack of reformed vesicles impaired recycling and eventually slowed down exocytosis as a secondary effect.
Importantly, the researchers found that a drug called nocodazole, which blocks new microtubule assembly, prevented injected tau from impairing endocytosis.
The next step for the researchers was to figure out exactly how an excess of microtubules caused a block of endocytosis.
While searching for a link between microtubules and endocytosis, the team realized that dynamin, a large protein that cuts off vesicles from the surface membrane at the final step of endocytosis, was actually discovered as a protein that binds to microtubules, although little is known about the binding site.
When the scientists fluorescently labeled tau, microtubules and dynamin, they found that presynaptic terminals that had been injected with tau showed an increase of bound dynamin, preventing the protein from carrying out its role in endocytosis.
Finally, the team created many peptides with matching sequences of amino acids to parts of the dynamin protein, to see if any of them could prevent dynamin from binding to the microtubules, and therefore rescue the signaling defects caused by tau protein. When one of these peptides, called PHDP5, was injected along with tau, endocytosis and synaptic transmission remained close to a normal level.
Moving forward, the researchers plan to test this peptide in Alzheimer mouse models that have increased levels of soluble tau. These mice lose their ability to learn and form new memories around 6-8 months old, and the team hope that the peptide can prevent or reverse this memory impairment.
"To this end, we must modify PHDP5 so that it can penetrate the blood brain barrier. If this peptide works in these mice models, it can serve as an effective therapeutic tool for Alzheimer's disease," said Prof. Takahashi.
More information: Tetsuya Hori et al, Microtubule assembly by tau impairs endocytosis and neurotransmission via dynamin sequestration in Alzheimer's disease synapse model, eLife (2022). DOI: 10.7554/eLife.73542