New genetic culprit suspected in the onset of pancreatic cancer
. DOI: 10.1016/j.celrep.2022.111623")
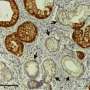
New research out of VCU Massey Cancer Center points to the inactivation of a previously unidentified gene as a likely culprit in the development of pancreatic cancer. Recently published in Cell Reports, the findings could alter the scientific understanding of this deadly disease and inform the establishment of novel treatments.
The findings hold implications for the targeted treatment of pancreatic ductal adenocarcinoma (PDAC), which accounts for the vast majority of all pancreatic tumors and is the fourth-leading cause of cancer-related deaths worldwide. Most patients are diagnosed at an advanced stage when the disease is already inoperable and there are no effective therapies.
Extensive research has demonstrated that mutations in the KRAS gene play an enormous role in the formation and growth of pancreatic cancer. Approximately 85-90% of all pancreatic tumors have a KRAS mutation.
"Given its high incidence at very early stages of the disease, mutational activation of KRAS has been hypothesized as the key genetic driver for pancreatic cancer," said study corresponding author Azeddine Atfi, Ph.D., leader of the Cancer Biology research program who holds the Mary Anderson Harrison Distinguished Professorship in Cancer Research at Massey.
However, there is a substantial fraction—around 10–15%—of PDAC tumors that harbors an offset of the conventional KRAS mutation, characterized as "wild-type KRAS." This suggests that in many cases alternate genetic drivers are responsible for the development of cancer.
Atfi's new research suggests that the inactivation of NF1—a gene known as Neurofibromin-1 that holds natural tumor-suppressing functions—could be instrumental in the onset of pancreatic cancer, either in tandem with KRAS, bolstering its cancer-driving properties, or even before any mutations occur in the KRAS gene, in partnership with TP53, the most inactivated tumor suppressor gene in human malignancies.
Atfi and his collaborators determined that removing NF1 in mice without KRAS mutations directly resulted in the early developmental stages of pancreatic tumors, but also enhanced the cancer-driving function of KRAS in mice with mutations.
"We found that genetic inactivation of NF1 dramatically accelerates the formation and progression of KRAS-mediated pancreatic cancer," said Atfi, who is also a professor in the Department of Biochemistry and Molecular Biology at the VCU School of Medicine. "This study raises the provocative possibility that targeting NF1 in mutant KRAS-bearing pancreatic tumors might create vulnerabilities that could be exploited for therapeutic advantage."
Additionally, the researchers observed a strong association between NF1 and p53, another protein widely known for its tumor-suppressing functions. They found that the simultaneous inactivation of both NF1 and p53 directly correlated to pancreatic cancer growth, regardless of any mutations in the KRAS gene.
"The notion that the combined inactivation of NF1 and p53 represents an alternative initiating event in PDAC provides an unprecedented platform for the future identification of novel targeted treatment options for pancreatic cancer," Atfi said.
More information: Gopalakrishnan Ramakrishnan et al, NF1 loss of function as an alternative initiating event in pancreatic ductal adenocarcinoma, Cell Reports (2022). DOI: 10.1016/j.celrep.2022.111623