This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Possible genetic basis and mouse model found for severe nonalcoholic fatty liver disease

A mutant or damaged gene may be a cause of a severe, mysterious form of nonalcoholic fatty liver disease, University of Illinois Urbana-Champaign researchers have found. Mice and human liver cells lacking the SRSF1 gene show all the hallmarks of nonalcoholic steatohepatitis, also known as NASH, the researchers discovered.
The unique mouse model captures all three hallmarks of excess fat, inflammation and scarring in the liver, opening the doors to better understanding and development of treatments for NASH, said study leader Auinash Kalsotra, a biochemistry professor at Illinois. The researchers published their results in the journal Nature Communications.
Nonalcoholic fatty liver disease affects an estimated 25% of Americans, according to the National Institutes of Health, and up to a third of patients will develop inflammation and scarring as well, falling under the NASH classification.
"It's not really clear why certain people end up developing NASH," Kalsotra said. "So far, there haven't been any genes that have been directly linked to NASH. It's been thought of as a progression: After the liver is injured or it becomes fatty, if you don't check it, it's going to progress toward NASH. But not everybody with fatty liver develops NASH and it seems random who does, so it's very confusing as to why this happens."
The other problem making NASH research difficult is the lack of good animal or cellular models for it, Kalsotra said. While some models incorporate one or two of the symptoms, and then other damage inflicted to mimic what is seen in NASH patients, they don't give an accurate portrayal of the disease.
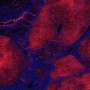
Kalsotra's group began studying SRSF1, a protein that assists in splicing RNA in the cell, to study its splicing activity. They bred a line of mice that were lacking the gene to learn about its splicing activity. The mice soon spontaneously developed all the symptoms of NASH.
"We were very excited to see this, as we thought, this could be a model for the disease. But first, we had to figure out the mechanisms of why SRSF1 was connected to liver disease," said graduate student Waqar Arif, the first author of the study.
 Overlap of differentially expressed genes from RNA-seq (FDR")
The researchers found that the mechanism was not related to SRSF1's splicing activity, as first thought, but because it protected against DNA damage in the liver during the process of DNA unwinding so its code could be transcribed into RNA. Without the SRSF1 protein, liver cells began to shut down.
"What happened was the cell wanted to die because of the extensive damage that was happening to the DNA. But it didn't have the energy for regular programmed cell death processes. The damage was so severe that everything just shut down," said Kalsotra, who also is affiliated with the Carl R. Woese Institute of Genomic Biology and the Cancer Center at Illinois.
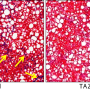
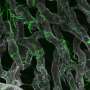
It was the opposite of the pathway that had been assumed, Kalsotra said. Instead of excess fat leading to cell damage, the cell damage came first, and fat built up as a result of the cells not being able to perform their function of packaging up digested fats. Inflammation increased as the immune system tried to clear away dead and dying cells, and then fibrosis appeared as scars formed over patches of dead tissue to try to hold the liver together. The researchers saw this same sequence in both the mice without SRSF1 and in human liver cell lines depleted of SRSF1.
"These findings tell us that the genome of the liver cells has to be properly maintained," Kalsotra said. "Liver cells are encountering toxins and DNA damage more than any other cells in our body. This study illustrates that when liver cells encounter excessive DNA damage, these NASH symptoms develop. SRSF1 plays a protective role against that. It acts as a guardian of the liver genome."
The researchers hope the mice will aid other researchers studying NASH, both in terms of understanding the disease and in exploring potential treatment targets.
"We want to see whether there are certain conditions where SRSF1 has been inactivated in some way in human livers or other animals," Kalsotra said. "Maybe there are some toxins or medicines or other environmental pollutants, for example, that could cause inactivation of the SRSF1 gene. Figuring out the triggers that cause silencing of SRSF1 could point to treatment targets, and whether things like anticancer drugs that prevent DNA damage could be applied here. It's interesting to note that many NASH patients develop cancer as well. Is that linked to the DNA damage?"
The researchers also plan to explore whether other genes protect DNA in this way, and whether damage to any of them could cause NASH pathology as well.
More information: Wagar Arif et al, Splicing factor SRSF1 deficiency in the liver triggers NASH-like pathology and cell death, Nature Communications (2023). DOI: 10.1038/s41467-023-35932-3. www.nature.com/articles/s41467-023-35932-3