March 27, 2023 report
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Comprehensive testing technique for post-translational modification drug interactions
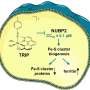
 The decryptM workflow comprises 7 steps: 1. Treatment of cells with increasing concentrations of a drug of interest and for a certain period of time. 2. Proteins are extracted from drug-treated cells. 3. Proteins are digested with trypsin. 4. Tryptic peptides are stable isotope labeled by tandem mass tags (TMT; one specific TMT reagent for each drug dose or time point). 5. Enrichment of a post-translational modification by immunoprecipitation (acetylation, ubiquitinylation) or immobilized metal affinity chromatography (IMAC). 6. TMT labeled samples are combined and subjected to LC-MS/MS analysis on an Orbitrap mass spectrometer. 7. Peptides and proteins are identified from MS2 spectra and relative quantification of peptides and proteins is performed on the basis of the TMT reporter intensities both using the software MaxQuant. A pipeline of custom decryptM scripts are used for fitting dose-response curves, filtering for significant drug perturbations, and creating different visualizations. Credit: Science (2023). DOI: 10.1126/science.ade3925")
A research team of 37 scientists, led by the Technical University of Munich, has developed a method to interrogate drug-induced post-translational modifications (PTMs) in a time and dose-dependent way.
The study, published in the journal Science, details an approach that more closely mimics what is happening in cells when drug proteins interact with proteins in a cell. Post-translational modifications (PTMs) are processing events that affect the structure and dynamics of proteins.
Because the structure of a protein is specific to its function, the modifications can significantly alter biological processes, which is how most drugs are designed to operate. While the drug/cell protein interactions are generally understood, exactly how this interaction takes place over the course of a treatment (time and dose-dependent) is an understudied aspect.
The proteomic assay method, called decryptM, involves treating cells with increasing concentrations of a drug and quantifying thousands of drug-PTMs to reveal target engagement and drug mechanism of action.
For the study, decryptM profiling was applied to 31 cancer drugs in 13 cell lines. The data derived from the method represents 1.8 million quantitative cellular drug assays with dose-response curves, with 124,660 regulated phosphopeptides detected on 11,982 proteins, 9,173 ubiquitinylated peptides detected on 3,006 proteins and 2,478 regulated acetylated peptides detected on 1,377 proteins.
Most PTMs were not regulated by most drugs, which is valuable information for understanding which pathways are being used or perhaps missed by each medicine.
In the case of two proteasome inhibitor drugs, bortezomib and carfilzomib, decryptM data revealed that drug effects became more potent over time and suggested the involvement of increased regulated phosphorylation sites as a possible mechanism.
The study also found that different histone deacetylase inhibitors (anti-cancer agents) have different activation times and that specific targets of the drugs were more potent than others. This type of information could greatly benefit researchers looking to improve the effectiveness of existing drugs.
Researchers also looked at multiple breast cancer cell types with decryptM profiles. The signaling pathways in different cancer cells can diverge strongly, and decryptM profiling uncovered cell-line specific signatures of drug interaction. For example, a cancer drug regulated hundreds of phosphopeptides in two cell types tested but merely regulated five in another. This is precisely the sort of drug/cell type interaction information that a doctor or pharmaceutical researcher would want to know about.
The study highlights some limitations of the approach in its current form. There is difficulty in attributing the resulting data to a specific target if a drug can engage multiple targets with similar potency. Still, this makes decryptM profiles a powerful starting point for more experimentation.
The authors envision that decryptM profiles may serve to monitor and eventually predict drug responses in-vivo once sufficient drugs and cell systems have been analyzed. Additionally, matching decryptM profiles of cancer drugs with cancer patient PTM profiles may become an important tool in personalized and evidence-based treatment recommendations.
More information: Jana Zecha et al, Decrypting drug actions and protein modifications by dose- and time-resolved proteomics, Science (2023). DOI: 10.1126/science.ade3925
© 2023 Science X Network