June 30, 2023 report
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Xeroderma pigmentosum study tests artificial antisense oligonucleotides as therapeutic
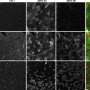
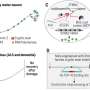
 Estimated structures of pre-mRNA products resulting from the ERCC4/XPF intron variants. Cryptic intron fragments identified in the patients’ mRNA are shown in blue lines. (B) cDNA sequences of the ERCC4/XPF exons 1 to 2 boundary in 48BR (normal) and XP43NG (XP-F). The 5′ cryptic intron 1 fragment is shown in blue letters. (C) A cDNA sequence of the ERCC4/XPF exon8–intron9 boundary in XP2YO (XP-F). The cryptic intron 8 fragment is shown in blue letters. Arrowhead indicates the variant position. (D) Digital quantitative PCR (digital qPCR) detected the reduction of ERCC4/XPF expression and the aberrant splicing product of intron 1 in XP43NG (filled bars, PCR amplifying the ERCC4/XPF exons 1 to 2 boundary; gray bars, PCR amplifying the 5′ cryptic fragment of ERCC4/XPF intron 1; open bars, a control PCR product of the TBP gene). (E) Digital qPCR detected the reduction of ERCC4/XPF expression in XP3YO (XP-F) (filled bars, PCR amplifying the ERCC4/XPF exons 1 to 2 boundary; hatched bars, exons 7 to 8 boundary; gray bars, exons 8 to 9 boundary; open bars, TBP). (F) Immunoblotting of the XPF protein. wild type and ΔERCC4/XPF, wild type and ERCC4/XPF-deficient HeLa cells; 48BR, normal; XP24BR, XP-F control; [XP136KO, XP37NG, XP43NG, XP101OS, XP97NG, XP165KO, XP103NG, XP4NG, XP48NG, XP90NG, XP95NG, XP18NG, XP133KO, XP23OS, XP96NG, XP2YOSV40, and XP3YO], the Japanese XP-F cases. b-actin (ACTB) as a loading control. XPF-upper bands represent the stop-loss product, p.*917Rext*83. Credit: Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2217423120")
Genetic researchers at Nagoya University, Japan, have delved into the genetic underpinning of a rare skin condition affecting children that is unusually common in Japan.
In the paper, "Deep intronic founder mutations identified in the ERCC4/XPF gene are potential therapeutic targets for a high-frequency form of xeroderma pigmentosum," published in PNAS, the team finds a potential therapeutic target for the disorder with artificial antisense oligonucleotides.
Xeroderma pigmentosum (XP) is a rare genetic disorder that can cause heightened sensitivity to sunlight and an increased risk of skin tumors due to a deficiency in the DNA repair system responsible for processing sunlight-induced photolesions.
XP patients commonly experience severe skin problems, including photosensitivity, dry skin, pigmentation abnormalities, and a heightened risk of skin cancer. Some cases may also exhibit neurological symptoms.
The clinical manifestations of XP vary depending on the affected genes and types of mutations. While every form of XP exhibits some of the pathologies, XP-F patients can have most or all of the disease manifestations at once.
The worldwide prevalence of XP is rare at approximately 3 in a million. In Japan, the rates are much higher, 1 out of 22,000. Of XP cases, XP-F incidents are around 1% globally and 4% in Japan, making XP-F 66 times more common in Japan than the global average.
Partly because XP is such a rare disorder, it remains under-studied, and treatment options are limited. There is a need for improved understanding, diagnosis, and potential therapeutic targets for XP, especially for the rarest variants like XP-F.
The study was conducted on a Japanese XP cohort (cohort size identified only as "largest") and identified 17 XP-F cases, all of which had one of two ERCC4/XPF gene variants. The first variant is a Japanese founder mutation that accounts for approximately 10% of all Japanese XP cases and causes incorrect pre-mRNA splicing. The second mutation induces alternative polyadenylation.
Both mutations result in reduced ERCC4/XPF gene expression, leading to a significant reduction in XPF protein expression and DNA repair deficiency in patients' cells.
The team attempted to correct the abnormal splicing events in patient cell samples through artificial antisense oligonucleotides (ASOs). ASOs work by binding to specific mRNA sequences. They can induce degradation, modulation of splicing, prevention of translation, or in this case, interfere with alternative mRNA processing events, which can inhibit or upregulate the production of downstream proteins.
After treatment with ASOs, the XPF protein expression was recovered to the normal level, indicating that the ASOs efficiently restored the mRNA expression. The study demonstrates that antisense oligonucleotides specifically designed for these mutations can restore XPF protein expression and DNA repair capacity in cells from XP-F patients.
While ASOs are currently being developed and tested for a variety of genetically based diseases, the application in the current study illustrates how genetic studies can lead the way in finding therapeutic targets for even the rarest of diseases.
More information: Chikako Senju et al, Deep intronic founder mutations identified in the ERCC4 / XPF gene are potential therapeutic targets for a high-frequency form of xeroderma pigmentosum, Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2217423120
© 2023 Science X Network