This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Data-processing tool could enable better early stage cancer detection
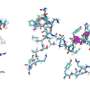
 and the scDNA-seq absolute copy numbers (or their log-transformed values) that are supposedly obtained from the same tissue. Distinct clones are distinguished by color saturation. b Given the gene expression and copy number matrices (intensity of pixel colors is proportional to copy number/gene expression values), MaCroDNA identifies the assignment of the scRNA-seq cells (dark orange circles) with gene expression values to the scDNA-seq cells (dark blue circles) with copy number values. When the number of the scRNA-seq cells is higher than the number of the scDNA-seq cells (as in the above example), MaCroDNA infers the correspondence between the cells (shown by pink arrows) by solving a series of maximum weighted bipartite matching problems (in this example, two steps are required). In the first step, only six scRNA-seq cells (equal to the number of the scDNA-seq cells) are assigned to the scDNA-seq cells such that no two scRNA-seq cells are paired with the same scDNA-seq cell, and the sum of the Pearson correlation coefficients between the pairs is maximized. c The scRNA-seq cells whose correspondences were identified in the last step are removed. Next, the remaining three scRNA-seq cells are assigned to the best scDNA-seq cells in a one-to-one fashion according to the same correlation-based criterion. d The clones are inferred from the scDNA-seq data using an algorithm of choice (blue, orange, and green bubble shapes represent the clones). e Given the cell-to-cell correspondences and the clonal assignment of the scDNA-seq cells, we can assign the scRNA-seq cells to the scDNA-seq clones. The clonal assignment of a scRNA-seq cell is that of its corresponding scDNA-seq cell. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-44014-3")
Cancers begin with abnormal changes in individual cells, and the ability to track the accumulation of mutations at the single-cell level can shed new light on the early stages of the disease. Such knowledge could enable more effective early detection and treatment options for patients as well as more accurate predictions of disease progression.
According to a paper in Nature Communications, a team of Rice University researchers led by Luay Nakhleh has developed a platform for integrating DNA and RNA data from single-cell sequencing with greater speed and precision than more recent, state-of-the-art technologies.
The method, mapping cross-domain nucleic acid or MaCroDNA, relies on a classical algorithm to identify matching pairs of data from DNA—the genetic blueprint of a cell—and RNA—a cell's instruction manual for protein assembly.
"Imagine you are given two large sets of photos of cars with the license plates and other identifying features blurred," said Mohammadamin Edrisi, a Rice Ph.D. student in computer science and lead author on the study.
"One set contains photos of the cars taken from the front, while the other set has photos of the back of the cars, and someone asks you to find the pairs of photos that belong to the same car. This is a metaphor for the problem we have tried to solve. The cars are cancer cells, and the two sets of photos are DNA and RNA data measurements."
In fact, the scenario that MaCroDNA is designed to address is more complex than that.
"In a typical cancer single-cell sequencing experiment, the DNA and RNA data sets are obtained from different cells in the tumor sample," said Nakhleh, the senior author of the study. "So the matching in such a scenario happens between cells that we know are not the same cells."
"To continue the analogy, think of each photo as being taken of the front or back of a different Toyota car, and we want to match pairs of photos that belong to a car of the same model—the front and back of a Toyota Camry, of a Toyota Corolla, etc. Different car models here are analogous to different clones within a heterogenous tumor, where each clone is expected to have very similar, yet not completely identical, DNA and RNA signatures across all cells within the clone."
Single-cell sequencing has developed significantly over the past decade, driving discovery across various fields of biology. This sequencing technique is an effective tool for studying how changes at the level of the genetic code impact cells' makeup or functioning, making it easier to track the types of transformations that turn a population of healthy cells into malignant tissue.
"Cancer cells demonstrate abnormal RNA patterns, and one of the reasons for that is DNA mutations," Edrisi said.
In their quest to identify the best tool for the task, the researchers tested a variety of methods against a real biological dataset with known matching DNA-RNA pairs.
"We tested the state-of-the-art method—named clonealign—and the other widely used methods using a real dataset with ground truth information for accuracy measurement," Edrisi said. "Interestingly, using this dataset was one of the novelties in our work. Previous studies relied on simulated data for accuracy measurements, even though there is no scientific consensus as to how to go about simulating such data."
Of the different machine learning technologies they tested, the researchers found that using a classical correlation coefficient and the maximum weighted bipartite matching algorithm yielded the most accurate results. In other words, MaCroDNA outperformed clonealign by a significant margin.
"The surprising part of our work was that using the classical correlation instead of clonealign's complicated formula and incorporating it in an algorithm from the 1950s led to the best accuracy we have ever witnessed," Edrisi said. "The lesson is that we should never judge an algorithm based on its complexity. Give it a shot, and make sure it is compared to the others in a fair setting."
The method is available for use in cancer research on the role of DNA-RNA dynamics in the emergence of cancer.
More information: Mohammadamin Edrisi et al, Accurate integration of single-cell DNA and RNA for analyzing intratumor heterogeneity using MaCroDNA, Nature Communications (2023). DOI: 10.1038/s41467-023-44014-3