This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Decoding the language of epigenetic modifications
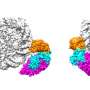
 to truncated histone cores (H3.1Δ1–31T32C or H4Δ1–28I29C, respectively). Note that this introduces H3T32C and H4I29C mutations that might affect protein binding to nearby modifications. Ligated histones were refolded into octamers and assembled into dinucleosomes using a biotinylated DNA containing two nucleosome-positioning sequences (di-601)47. For some experiments, CpG-methylated DNA (m5C) or H2A.Z were used. b, SNAP purifications. Modified nucleosomes were immobilized on streptavidin beads and incubated with nuclear extracts from HeLa S3 cells grown in isotopically light (R0K0) or heavy (R10K8) SILAC medium. c, Protein responses to modified nucleosomes. For each SNAP experiment, bound proteins were identified and quantified using MS, and the forward (x axis) and reverse (y axis) SILAC ratios (H/L ratio) were plotted on a logarithmic (log2) graph. d, A library of modified dinucleosomes. A header specifies the modification status of each nucleosome. Nucleosomes are arranged in columns, with the respective modifications displayed in rows. Modifications of specific lysine residues in histone H3 and H4 and the presence of DNA methylation (meCpG) or H2A.Z are color coded as indicated. Nucleosomes are ordered to imitate clustering by increasingly active chromatin states. Monometh., monomethylation; PTMs, post-translational modifications. e, Visualization of protein binding responses to the 55 modified dinucleosomes profiled by SNAP. The log2[H/L] ratios for each protein in each SNAP experiment are shown as circles, with the right half representing the forward and the left half the reverse log2[H/L ratio]. Recruitment (red) and exclusion (blue) are indicated. The reverse H/L ratio was inverted to display both ratios on the same scale. Circle sizes denote the total MS1 peak intensities on a log10 scale. Credit: Nature (2024). DOI: 10.1038/s41586-024-07141-5")
Epigenetic changes play important roles in cancer, metabolic and aging-related diseases, but also during loss of resilience as they cause the genetic material to be incorrectly interpreted in affected cells. A major study by scientists at Helmholtz Munich, now published in Nature, provides important new insights into how complex epigenetic modification signatures regulate the genome. This study will pave the way for new treatments of diseases caused by faulty epigenetic machineries.
Our bodies are made up of hundreds of different cell types, each with its unique shape and function. The information on how to build an organism is stored in our DNA. However, while all our cells share the same DNA, they don't all read it in the same way.
So how does, for example, a liver or a brain cell know which instructions to follow? To make this possible, small chemical tags, so-called epigenetic modifications, are used. They act like flags and tell each cell which parts of the DNA to use and which ones to ignore.
Simple at first glance, this epigenetic regulation is much more complex, as there are many different modifications that can either be attached directly to our DNA or to so-called histone proteins. "Histones are small proteins around which our DNA is wrapped, and which thereby serve to package the genetic material," says study leader Dr. Till Bartke, deputy director of the Institute of Functional Epigenetics (IFE) at Helmholtz Munich.
"Depending on how the histones or the DNA are chemically modified, they can have different effects on the DNA and thereby control gene activity." Together, epigenetic modifications form what scientists call the epigenetic code, allowing cells to switch genes on or off according to their specific needs.
However, how these epigenetic modifications work together has remained a big puzzle. Finding out how this epigenetic code works is the focus of the research at the Institute of Functional Epigenetics led by Prof. Robert Schneider. "Our understanding of the complex interaction between our DNA and epigenetic mechanisms has now taken an important step forward with this groundbreaking study from our institute," Schneider says.
Cracking the epigenetic code in a test tube
To decipher the epigenetic code, Till Bartke and co-workers developed a creative way to examine how different combinations of epigenetic modifications work together. They reconstructed many of these modifications in a test tube and carried out experiments to study how they interact with the proteins in our cells, using a combination of sophisticated biochemical and mass spectrometric methods.
"Epigenetic modifications usually act in cooperation with so-called epigenetic reader proteins that recognize them and promote downstream effects," explains Dr. Andrey Tvardovskiy, post-doctoral researcher and one of the first authors of the study. "Uncovering how epigenetic readers interpret such complex modification signatures is therefore key to understanding how our genome functions and how its misregulation can lead to human diseases."
For the first time, the researchers could see how different combinations of modifications are "read" and translated by the protein machinery in our cells.
Using newly developed AI approaches, they next set out to decode the language of epigenetic modifications. The researchers found that some constituents of the epigenetic code have a big impact, especially on stretches of the DNA that control gene activation, while others have a smaller effect. By putting together all this information, they managed to extract several fundamental rules of how our genetic material is organized and controlled inside our cells.
These insights are highly relevant for many scientists across different fields and are anticipated to catalyze many future discoveries. To ensure that their findings are as widely available as possible, the researchers built a website called the Modification Atlas of Regulation by Chromatin States that provides an intuitive interactive online resource to explore the results of their study.
"Since epigenetic modifications play crucial roles in everything our bodies do, from growing and learning to staying healthy, things go wrong when the modifications are misplaced or misread. Often, this causes diseases like cancer, developmental disorders, or mental disabilities," says Till Bartke.
"But epigenetic changes also accumulate throughout life and are affected by the environment, nutrition, and lifestyle—this can contribute to diseases such as diabetes and lead to deleterious effects of aging."
By understanding how epigenetic modifications work and what goes wrong in diseases, researchers at the IFE aim to develop new ways to treat these diseases and tackle adaptation to a changing environment.
More information: Till Bartke, Decoding chromatin states by proteomic profiling of nucleosome readers, Nature (2024). DOI: 10.1038/s41586-024-07141-5. www.nature.com/articles/s41586-024-07141-5