This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Study reveals key mechanisms of a rare form of epilepsy
. DOI: 10.7554/eLife.91204.1")
A study has identified a genetic mutation underlying a rare form of epilepsy, and reveals novel molecular and cellular mechanisms by which the disorder manifests in patients.
The research, published today as a Reviewed Preprint in eLife, is described by the editors as having fundamental significance as it substantially advances our understanding of KCNQ2 encephalopathy. They describe the methods used as state-of-the-art, providing a convincing strength of evidence.
"KCNQ2 encephalopathy is a rare neurodevelopmental disorder caused by variants in the KCNQ2 gene, which provides the recipe for a type of brain potassium channel," explains lead author Timothy Abreo, a student in the Genetics and Genomics Graduate Program at Baylor College of Medicine, Houston, Texas, US. "The disorder usually manifests as seizures beginning within days after birth, and developmental delays which are lifelong and without effective current treatment."
Newly identified variants in the KCNQ2 gene are hard to assess, because not every variant is disease-causing, and the reasons why some variants do lead to illness have been poorly understood.
The research team's study began when a female child, born in the US, experienced epileptic seizures that recurred throughout the first month of life. Genetic testing revealed a single amino acid change—glycine 256 changed to tryptophan (G256W)—on one copy of the KCNQ2 gene, which was previously unknown and classified as of uncertain clinical significance.
The team's study carefully characterized the clinical and developmental history of the young patient. In infancy, developmental delays became apparent, and she subsequently was diagnosed with cortical visual impairment and autism spectrum disorder. Now 12 years old, she is able to use an adaptive communication device to make requests, and use modified sign language. The researchers followed the scientific literature and later found three other individuals in Asia and the EU with the same G256W variant and symptoms similar to the US patient. This observational study component allowed the team to specify the features to be modeled in the laboratory.
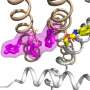
The team examined the structure and pathogenicity of the G256W variant using molecular, cellular, and in vivo approaches. They modeled the structural consequences of the G256W variant based on reported cryo-electron microscopy structures of the KCNQ2 channel and other similar channel proteins.
Like other potassium channels, KCNQ2 contains three critical regions: the turret, the pore helix, and the selectivity filter. Disease-causing variants tend to lie in residues near the selectivity filter, due to its crucial role enabling ions to flow. However, the G256 amino acid lies relatively far from the selectivity filter, which would typically indicate that a variant could be less likely to cause illness.
The team uncovered evidence that G256 helps form a network of hydrogen bonds that serve to stabilize the structure of the pore-forming region. This provided a novel hypothesis for why an amino acid mutation so distant from the selectivity filter might be pathogenic.
Next, Abreo and colleagues conducted a series of experiments in which they introduced the G256W variant into cells alongside normal KCNQ2 and KCNQ3 genes. They measured electrical activity in the cells and observed that the presence of the G256W variant suppressed the function of the normal gene, and reduced the ion current generated by the cells.
Finally, they used Crispr/Cas9 gene editing techniques and generated mice carrying the same G256W variant. Mice with G256W showed alterations in the distribution of KCNQ2 and KCNQ3 proteins within the brain, specifically in a region called the hippocampus that plays essential roles in learning and memory. They found that in the mouse models, KCNQ2 and KCNQ3 proteins were shifted from their usual location in axon initial segments to the neuronal somata.
Other experiments showed that the levels of KCNQ2 protein were decreased by roughly 50%, and the neurons in the CA1 region of the hippocampus showed heightened excitability, potentially indicating greater seizure susceptibility. Indeed, newborn and older G256W mice had spontaneous seizures that led to premature mortality not seen in control mice.
"Taken together, our study reveals that the presence of KCNQ2 G256W variants affects both molecular and cellular aspects of KCNQ channel activity, including their ion-carrying capacity, expression, and placement within cells," explains senior author Edward Cooper, Associate Professor of Neurology, Neuroscience, and Molecular and Human Genetics at Baylor College of Medicine. "These changes likely combine to drive the pathogenicity of the variant, and its clinical symptoms."
"Our results may be broadly applicable because the majority of KCNQ2 encephalopathy patients share variants near the selectivity filter," adds Abreo.
The US child's parents, who are also co-authors of the paper, played a crucial, 10-year long role in the study, sharing their history, fostering contact between clinicians and laboratory scientists, and helping Dr. Cooper build the multi-institutional lab team that generated and analyzed the mice.
"As parents and leaders of a parent-led foundation representing the global impacts of KCNQ2, we are excited to see these studies unlocking new mysteries of the disorder. Such progress is the path to better patient outcomes and precision medicine therapies," said Jim Johnson, father of the affected US child and President of KCNQ2 Cure Alliance*.
The authors encourage further research into this area to extend their findings. In particular, they highlight that, although mice with the G256W variant show spontaneous convulsive seizures and seizure-related deaths, they do not exhibit the very early onset and high frequency seen in humans with the same variant.
"This difference merits more investigation," they wrote. In addition, "selective KCNQ2 openers, which counter the effects of G256W in vitro as shown in our study, should be tested in mutant animals and if effective, in humans with KCNQ2 disease-causing variants," says Dr. Cooper.
More information: Timothy J. Abreo et al, Plural molecular and cellular mechanisms of pore domain KCNQ2 encephalopathy, eLife (2024). DOI: 10.7554/eLife.91204.1