This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Filling in genomic blanks for disease studies works better for some groups than others

Understanding how genetics affect health is an essential first step toward treating and preventing a host of diseases. New knowledge often comes from genome-wide association studies identifying variations in the genetic code linked with conditions such as cancer and autoimmune disease. The more people's DNA and health histories that are examined in such research, the more likely genetic and biological insights can be garnered.
However, cost can be a major barrier: Comprehensively sequencing one person's genome costs about $500 to $1000, a price point often infeasible when applied to several tens of thousands of study participants. So instead, researchers generally focus on key spots where the genetic code tends to vary among different individuals, through genotyping, which costs about $100 per participant. A statistical method called genotype imputation then helps them fill in the genetic blanks based on existing reference panels of fully sequenced genomes.
A new Keck School of Medicine of USC study appearing in the American Journal of Human Genetics identifies a disparity in how well imputation works for different populations.
The researchers found that the technique holds up nicely for well-represented groups with European ancestry, as well as for African Americans and Latinos, who have been the subject of recent, concerted efforts to increase representation in sequencing reference panels. However, the researchers found that imputation is far less reliable for other groups, generally doing worse for populations farther away from Europe, except for Africa and Latin America.
"These global populations are not being imputed as well, meaning that we have a lot more error in filling in missing parts of the genome," said corresponding author Charleston Chiang, Ph.D., associate professor of population and public health sciences and associate director at the Keck School of Medicine's Center for Genetic Epidemiology. "That means the analysis using these imputed data doesn't work as well. And because researchers filter based on the reliability of imputation, we end up having data for diverse populations with more errors and more holes, leading to less effective study designs."
Reaching outside of a health science field to examine inequities
Chiang notes that the uniqueness of this study lies in the breadth of the study, where the team evaluated over a hundred global populations for issues with imputation. This has not been previously demonstrated because of the general lack of diversity of available cohorts as well as in reference panels of fully sequenced genomes. This presented a hurdle for understanding how well diverse groups fare with imputation in genetic epidemiology studies.
So the research team took a unique approach, borrowing genetic datasets from population genetics, a related field focused on understanding the history and evolution of a wide variety of populations, with less of a focus on disease.
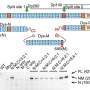
In all, the scientists combined genomic sequencing data from 23 studies including more than 43,000 people from 123 distinct global populations. They matched each population with a control group of European ancestry and used a standard metric that doesn't require full genomic sequences—which is normally the case in genome-wide association studies—to compare the reliability of imputation.
Imputation for populations based in places such as Papua New Guinea, Thailand, Vietnam and Saudi Arabia was substantially less accurate than for populations of European descent. Chiang and his colleagues also plotted the relative reliability of imputation for different groups on a world map that is available online. Imputation for populations based in Asia, Australia, New Zealand and the Pacific Islands generally showed less accuracy.
The team also compared the main metric for the reliability of imputation used to arrive at these findings with a better metric that only works when full sequencing data is available. They found that the main metric is biased so that it overestimates the accuracy of imputation for populations other than people of European ancestry. This suggests that the flaws in imputation are more serious still than indicated by the researchers' results.
Potential steps to make genome-wide association studies more equitable
The solution for the disparity highlighted in the study is straightforward, yet far from simple to achieve.
"We need to sequence more, and be more inclusive in the individuals who participate in studies," said Chiang, who also holds an appointment in quantitative and computational biology at USC Dornsife College and is a member of USC Norris Comprehensive Cancer Center.
One promising sign is that genomic sequencing has become more affordable in recent years and is expected to continue to do so. But cost isn't the only concern that must be addressed. Efforts are needed to earn the trust from diverse communities so they are not hesitant to participate.
In some cases, more diversity can complicate genome-wide association studies, particularly in smaller studies, even confounding their findings if the diversity is not properly accounted for or characterized. This creates pressure for scientists to exclude a smaller subset of populations in their data and choose from groups with more members.
Chiang advocates for a sort of balance.
"As the studies get bigger and bigger, the way that scientists view and analyze these data needs to evolve toward looking at genetic ancestry as more of a continuum," he said. "If we can start to view everyone as related and branching off the same genetic tree at different places, according to their history, we can incorporate more people and more diversity.
"Of course, there are valuable reasons to study discrete populations," he continued. "Group identity can be useful to maintain, for example when studying the social determinants of health that affect what people experience in their daily lives. We need to continue studying particular populations in isolation, but in the long term, we need to be able to reconcile between the two approaches."
The study's first author, USC undergraduate Jordan Cahoon, hopes that by beginning to quantify disparities baked into genome-wide association studies, the team's work will influence future solutions.
"It's important to understand the weaknesses in the field in terms of equity and fairness," said Cahoon, a graduating senior majoring in computer science at the USC Viterbi School of Engineering. "I'm hoping that this study will be a good resource for scientists, so they can see how well the populations they're sequencing are doing in comparison to others."
Other co-authors are Xinyue Rui, Echo Tang, Christopher Simons, Jalen Langie, Minhui Chen and Ying-Chu Lo, all of USC.
More information: Jordan L. Cahoon et al, Imputation accuracy across global human populations, The American Journal of Human Genetics (2024). DOI: 10.1016/j.ajhg.2024.03.011