This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
After 25 years, researchers uncover genetic cause of rare neurological disease

Some families call it a trial of faith. Others just call it a curse. The progressive neurological disease known as spinocerebellar ataxia 4 (SCA4) is a rare condition, but its effects on patients and their families can be severe. For most people, the first sign is difficulty walking and balancing, which gets worse as time progresses. The symptoms usually start in a person's forties or fifties but can begin as early as the late teens. There is no known cure. And, until now, there was no known cause.
Now, after 25 years of uncertainty, a multinational study led by Stefan Pulst, M.D., Dr. med., professor and chair of neurology, and K. Pattie Figueroa, a project manager in neurology, both in the Spencer Fox Eccles School of Medicine at University of Utah, has conclusively identified the genetic difference that causes SCA4, bringing answers to families and opening the door to future treatments. Their results are published in the journal Nature Genetics.
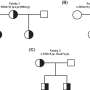
SCA4's pattern of inheritance had long made it clear that the disease was genetic, and previous research had located the gene responsible for a specific region of one chromosome. But that region proved extraordinarily difficult for researchers to analyze: full of repeated segments that look like parts of other chromosomes, and with an unusual chemical makeup that makes most genetic tests fail.
To pinpoint the change that causes SCA4, Figueroa and Pulst, along with the rest of the research team, used a recently developed advanced sequencing technology. By comparing DNA from affected and unaffected people from several Utah families, they found that in SCA4 patients, a section in a gene called ZFHX3 is much longer than it should be, containing an extra-long string of repetitive DNA.
Isolated human cells that have the extra-long version of ZFHX3 show signs of being sick—they don't seem able to recycle proteins as well as they should, and some of them contain clumps of stuck-together protein.
"This mutation is a toxic expanded repeat and we think that it actually jams up how a cell deals with unfolded or misfolded proteins," says Pulst, the last author on the study. Healthy cells need to constantly break down non-functional proteins. Using cells from SCA4 patients, the group showed that the SCA4-causing mutation gums up the works of cells' protein-recycling machinery in a way that could poison nerve cells.
Hope for the future
Intriguingly, something similar seems to be happening in another form of ataxia, SCA2, which also interferes with protein recycling. The researchers are currently testing a potential therapy for SCA2 in clinical trials, and the similarities between the two conditions raise the possibility that the treatment might benefit patients with SCA4 as well.
Finding the genetic change that leads to SCA4 is essential to develop better treatments, Pulst says. "The only step to really improve the life of patients with inherited disease is to find out what the primary cause is. We now can attack the effects of this mutation potentially at multiple levels."
But while treatments will take a long time to develop, simply knowing the cause of the disease can be incredibly valuable for families affected by SCA4, says Figueroa, the first author on the study. People in affected families can learn whether they have the disease-causing genetic change or not, which can help inform life decisions such as family planning.
"They can come and get tested and they can have an answer, for better or for worse," Figueroa says.
The researchers emphasize that their discoveries would not have been possible without the generosity of SCA4 patients and their families, whose sharing of family records and biological samples allowed them to compare the DNA of affected and unaffected individuals.
"Different branches of the family opened up not just their homes but their history to us," Figueroa says. Family records were complete enough that the researchers were able to trace the origins of the disease in Utah back through history to a pioneer couple who moved to Salt Lake Valley in the 1840s.
Since meeting so many families with the disease, studying SCA4 has become a personal quest, Figueroa adds. "I've been working on SCA4 directly since 2010 when the first family approached me, and once you go to their homes and get to know them, they're no longer the number on the DNA vial. These are people you see every day… You can't walk away. This is not just science. This is somebody's life."
More information: GGC expansion in ZFHX3 causes SCA4 and impairs autophagy, Nature Genetics (2024). DOI: 10.1038/s41588-024-01719-5. www.nature.com/articles/s41588-024-01719-5