Finding that links ALS/ataxia to cellular stress opens new approaches for treatment

Few treatments exist for neurodegenerative diseases that progressively rob a person's ability to move and think, yet the results of a new study could potentially open additional approaches for exploration.
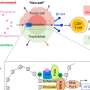
Scientists at University of Utah Health report for the first time that a protein, called Staufen1, accumulates in cells of patients suffering from degenerative ataxia or amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease. Depleting the protein from affected mice improved symptoms including motor function. These results suggest that targeting Staufen1 could have therapeutic potential in people. The research is published in Nature Communications.
"This is a completely new avenue for thinking about neurodegenerative diseases," says Stefan Pulst, M.D., Dr. Med., chair of Neurology at U of U Health and senior researcher on the study. "A protein that had never been known to be involved in neurodegeneration is now a great target for drug treatments."
Previously, researchers had not considered Staufen1 a culprit in neurodegenerative disease until they discovered its association with ataxia, a rare condition that causes patients to lose control of their movement. They found that Staufen1 binds Ataxin2, a protein that is both responsible for ataxia and a risk factor for ALS.
A role for Staufen1 in disease pathology became evident upon genetically depleting it from mice with an ataxia-like condition. The animals' condition improved at both physiological and molecular levels.
Beginning at 12 weeks of age, mice performed significantly better on a rotarod performance test, measuring the length of time the animals could walk or run on an accelerating spinning rod. In addition, the expression of a handful of proteins that had diminished in brain cells during disease reverted back to near-normal levels.
"Staufen was first discovered in the fruit fly and has been studied for 30 years, but it had never been connected to anything related to disease," says Pulst. "This is a novel finding." Future investigations will focus on determining whether drugs or therapies that reduce Staufen1 could be developed as treatments for multiple diseases.
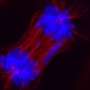
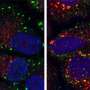
Beyond these applications, the biology of Staufen1 could reveal new clues about neurodegenerative disease. The protein accumulates with Ataxin2 and other proteins and RNAs in dense clusters called stress granules, a hallmark of ataxia, ALS and other conditions such as frontotemporal dementia. When Staufen1 was depleted from mice with ataxia, it not only improved the pathology of disease but also rid cells of stress granules.
While the precise role of stress granules is still an intensive area of study, they are believed to help cells weather stress caused by toxins or certain disease conditions, explains co-author Daniel Scoles, Ph.D., associate professor of Neurology at U of U Health. One function could be to prevent proteins from being made under suboptimal conditions.
The findings connect Staufen1 to the emerging concept that neurodegenerative diseases are linked to malfunctions in the way cells cope with cellular stress. One implication, says Scoles, is that Staufen1-targeted therapies could work against a number of disorders in which stress granules emerge, although it remains to be determined whether the aggregates themselves lead to disease.
"Our results put the stress granule in focus as a structure to target in disease," says Scoles.
More information: Sharan Paul et al, Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration, Nature Communications (2018). DOI: 10.1038/s41467-018-06041-3