Study uncovers mechanism by which tumor suppressor MIG6 triggers cell suicide

Death plays a big role in keeping things alive. Consider the tightly orchestrated suicide of cells—a phenomenon essential to everything from shaping an embryo to keeping it free of cancer later in life. When cells refuse to die, and instead multiply uncontrollably, they become what we call tumors. An intricate circuitry of biochemical reactions inside cells coordinates their self-sacrifice. Tracing that circuitry is, naturally, an important part of cancer research.
In a major contribution to that effort Dr. Ingvar Ferby, a Ludwig researcher based at Uppsala University in Sweden, led a team of researchers who have determined the outsize role a small protein named Mig6 plays in such processes. Their findings, reported online in the September 11 issue of Developmental Cell, unveil a conceptually novel mechanism for the biological regulation of cell suicide.
The studies focus on epithelial tissue in the mammary glands of mice. Though diverse in function, such epithelial linings—found on the surfaces of virtually every organ and gland in the body—generally consist of a stack of functionally distinct cells layered over a membrane that acts like a floor. The outermost cells of epithelia constantly die, slough off and are replaced. This tightly regulated process is known as epithelial homeostasis; where it breaks down, tumors often ensue.
To figure out what exactly Mig6 does, Ferby and his colleagues studied what happens in the mammary glands of mice engineered to lack the gene for that protein. His laboratory had previously shown that such mice tend to spontaneously develop tumors—suggesting that Mig6 somehow suppresses cancer. In the current study, he and his team noticed that tiny ducts in the mammary glands of these mice were clogged with aberrantly proliferating epithelial cells.
The question was why. To find out, Ferby's team took epithelial cells from mutant mice and their normal littermates and used them to conduct an elegant series of experiments. They discovered that Mig6 is the chief instigator of suicide in these cells.
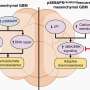
Mig6 was already known to dampen the signal of a secreted protein named epidermal growth factor (EGF) by binding to its receptor, and EGF plays an important role in maintaining epithelial homeostasis. But Ferby and his colleagues discovered that Mig6 really kicks into gear only after EGF is taken away. It drops off the EGF receptor and promptly latches on to an intracellular protein named cAbl. This activates cAbl, initiating a signaling cascade that reaches into the nucleus and turns on a key enzyme involved in cell suicide.
They next asked what keeps Mig6 from triggering cell death in the presence of EGF?
The answer, it seems, is yet another protein, Src, which is known to be activated by the EGF receptor. Ferby and his colleagues found that Src chemically modifies cAbl in a manner that prohibits its activation by Mig6. Conversely, when EGF is removed and its receptor becomes dormant, Src gets shut down—allowing Mig6 and cAbl to work together to induce cell suicide.
"In other words," explains Sarah Hopkins, a post-doctoral researcher at UCL (University College London) and the lead author of the study, "Mig6 is an intracellular sensor that detects the absence of a proliferative signal, such as EGF, and, in response, induces normal cell death."
Mig6 keeps the growth of epithelial cells dependent on EGF. This is notable because escape from such dependence is an early step in the generation of some tumors. Mig6 is known to be silenced in many human epithelial cancers, including those of the lung, pancreas, breast and skin.
"The findings from this study may have an impact on how we evaluate drugs that inhibit cAbl," says Ferby. "These therapies are designed to target a mutated version of cAbl that promotes cancer. But by disabling healthy cAbl, they may well interfere with a protein essential to Mig6's work inside the cell."
And that work, as these findings suggest, is clearly very important.
More information: 'Mig6 is a sensor of EGR receptor inactivation that directly activates c-Abl to induce apoptosis during epithelial homeostasis,' Sarah Hopkins, Emma Linderoth, Oliver Hantschel, Paula Surarez-Henriques, Giulia Pilia, Howard Kendrick, Matthew J. Smalley, Giulio Superti-Furga, Ingvar Ferby, Developmental Cell, online publication 11 September 2012.