Neuron-damaging mechanism discovered in mouse model of inherited amyotrophic lateral sclerosis
New research uncovers what may be a primary neuron-damaging insult that occurs in an inherited form of a devastating neurodegenerative disorder. The study, published by Cell Press in the August 26th issue of the journal Neuron, describes a critical mechanistic link between a mutant protein and disease pathogenesis in an animal model of amyotrophic lateral sclerosis (ALS).
ALS is a disease that attacks the neurons in the brain and spinal cord that control voluntary movement. There is no cure for ALS, which typically develops in adults and is characterized by a progressive paralysis that often leads to death within three to five years of diagnosis. About 10% of ALS cases are inherited and a portion of those are attributed to mutations in the gene for a protein called SOD1. However, the exact mechanism that links mutant SOD1 with motor neuron degeneration has not been established.
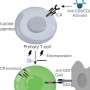
Malfunction of mitochondria, tiny intracellular energy-producing structures, has also been implicated in ALS pathology. "Previous research using rodent models and human samples showed that mutant SOD1 is associated with the outer surface of mitochondria in affected but not unaffected tissues," explains senior study author, Professor Don W. Cleveland from the University of California, San Diego. "Further, the voltage-dependent anion channel (VDAC1), which is also present in the outer membrane of mitochondria and controls communication between the mitochondria and the rest of the cell, has also been linked with cell death."
Professor Cleveland and colleagues built on these earlier observations and discovered that mutant SOD1 interacted with VDAC1 in the spinal cord of animals expressing mutant SOD1, and that this interaction disrupted VDAC1 function. Inhibition of VDAC1 function is known to decrease cellular energy production and drive formation of damaging reactive oxygen species.
The researchers went on to show that mutant SOD1-driven VDAC1 inhibition was seen in spinal cord mitochondria from mutant SOD1 expressing animals before symptoms developed and increased in severity during disease progression. Importantly, reduced VDAC1 activity accelerated the onset of fatal paralysis in ALS mice.
"Our evidence demonstrates that reduced VDAC1 function and correspondingly reduced mitochondrial function are direct components of intracellular damage from mutant SOD1," says Professor Cleveland. "The finding that VDAC1 is a target for mutant SOD1 within the nervous system provides important insight into the mechanism underlying premature degeneration and death of motor neurons."
More information: Israelson et al.: “Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS.” Publishing in Neuron, August 26, 2010.