Epigenomic findings illuminate veiled variants
Genes make up only a tiny percentage of the human genome. The rest, which has remained measurable but mysterious, may hold vital clues about the genetic origins of disease. Using a new mapping strategy, a collaborative team led by researchers at the Broad Institute of MIT and Harvard, Massachusetts General Hospital (MGH), and MIT has begun to assign meaning to the regions beyond our genes and has revealed how minute changes in these regions might be connected to common diseases. The researchers' findings appear in the March 23 advance online issue of Nature.
The results have implications for interpreting genome-wide association studies – large-scale studies of hundreds or thousands of people in which scientists look across the genome for single "letter" changes or SNPs (single nucleotide polymorphisms) that influence the risk of developing a particular disease. The majority of SNPs associated with disease reside outside of genes and until now, very little was known about the functions of most of them.
"Our ultimate goal is to figure out how our genome dictates our biology," said co-senior author Manolis Kellis, a Broad associate member and associate professor of computer science at MIT. "But 98.5 percent of the genome is non-protein coding, and those non-coding regions are generally devoid of annotation."
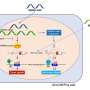
The term "epigenome" refers to a layer of chemical information on top of the genetic code, which helps determine when and where (and in what types of cells) genes will be active. This layer of information consists of chemical modifications, or "chromatin marks," that appear across the genetic landscape of every cell, and can differ dramatically between cell types.
In a previous study, the authors showed that specific combinations of these chromatin marks (known as "chromatin states") can be used to annotate parts of the genome – namely to attach biological meaning to the stretches of As, Cs, Ts, and Gs that compose our DNA. However, many questions remained about how these annotations differ between cell types, and what these differences can reveal about human biology.
In the current study, the researchers mapped chromatin marks in nine different kinds of cells, including blood cells, liver cancer cells, skin cells, and embryonic cells. By looking at the chemical marks, the researchers were able to create maps showing the locations of key control elements in each cell type. The researchers then asked how chromatin marks change across cell types, and looked for matching patterns of activity between controlling elements and the expression of neighboring genes.
"We first annotated the elements and figured out which cell types they are active in," said co-senior author Bradley Bernstein, a Broad senior associate member and Harvard Medical School (HMS) associate professor at Massachusetts General Hospital (MGH). "We could then begin to link the elements and put together a regulatory network."
Having pieced together these networks connecting non-coding regions of the genome to the genes they control, the researchers could begin to interpret data from disease studies. The team studied a large compendium of genome-wide association studies (GWAS), looking to characterize non-coding SNPs associated with control regions in specific cell types.
"Across 10 association studies of various human diseases, we found a striking overlap between previously uncharacterized SNPs and the control region annotations in specific cell types," said Kellis. "This suggests that these DNA changes are disrupting important regulatory elements and thus play a role in disease biology."
The researchers confirmed the reliability of their approach by showing that SNPs were associated with the appropriate cell types. For example, SNPs from autoimmune diseases such as rheumatoid arthritis and lupus sit in regions that are only active in immune cells, and SNPs associated with cholesterol and metabolic disease sit in regions active in liver cells. While more in-depth, follow-up studies will be needed to confirm the biological significance of these connections, the current study can help guide the direction of these investigations.
"GWAS has identified hundreds of non-coding regions of the genome that influence human disease, but a major barrier to progress is that we remain quite ignorant of the functions of these non-coding regions," said David Altshuler, deputy director at the Broad and an HMS professor at MGH, who was not involved in the study. "This remarkable and much-needed resource is a major step forward in helping researchers address that challenge."
SNPs in the non-coding regions of the genome may have subtler biological effects than their counterparts that arise in genes because they can influence how much protein is produced. The researchers mainly focused on SNPs in enhancer regions, which help boost a gene's expression, and their network connections to regulators that control them and genes that they target. Follow-up efforts can then focus on specific pieces of this network that could be targeted with drugs.
The team involved in this study hopes to expand its analysis to include many other cell types and map additional marks to expand their networks beyond enhancer regions. In the meantime, researchers involved in genome-wide association studies will be able to use the maps from this project to analyze non-coding SNPs in a new light.
"These maps can be used to come up with hypotheses about how the variants themselves are working and which ones are causal," said Bernstein. "This resource now goes back to the GWAS community, which can use the maps to form and test new functional models."
More information: Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. doi:10.1038/nature09906