Study links Fragile X Syndrome proteins and RNA editing mistakes at nerve-muscle junction
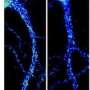
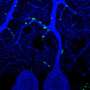
. Credit: Balpreet Bhogal, Perelman School of Medicine, University of Pennsylvania")
The most common form of heritable cognitive impairment is Fragile X Syndrome, caused by mutation or malfunction of the FMR1 gene. Loss of FMR1 function is also the most common genetic cause of autism. Understanding how this gene works is vital to finding new treatments to help Fragile X patients and others.
Researchers from the Perelman School of Medicine at the University of Pennsylvania, and colleagues from Brown University, have identified the FMRP protein (encoded by FMR1) as a key player in RNA editing, a process in which the working copies made from DNA, called messenger RNAs, are chemically altered after being transcribed from the genome. Their findings were published online this week in Nature Neuroscience.
Since RNAs are used as the instructions to make proteins, mistakes in RNA editing at the neuromuscular junction (NMJ), the site at which motor neurons innervate muscle, may cause problems in nerve function. Previous work at Penn and several other institutions strongly suggested the role of FMRP to be in regulating the translation of certain types of RNA at the synapse, the space between two nerves, or between nerves and muscles.
"Most of the field has been focused on looking at FMRP interacting with specific RNAs and how it regulates their translation at the synapse," states lead author Thomas A. Jongens, PhD, associate professor of Genetics at Penn. "Here we've tapped into identifying a function that FMRP has in regulating another process called RNA editing that is important in regulating neural activity." In RNA editing, the information encoded by DNA into an RNA molecule is altered, thus affecting the functioning of the proteins encoded by that RNA.
"This work elegantly links the Drosophila FMR1 gene to both an RNA-editing pathway and the architecture of the neuromuscular junction synapse," says Donna Kransnewich, PhD, who oversees grants focused on mechanisms of human genetic disorders at the National Institute of General Medical Sciences of the National Institutes of Health. "These exciting findings bring us closer to understanding the molecular basis of Fragile X syndrome, the most common inherited intellectual disability, and highlight the value of basic science research in uncovering the underlying causes of human disorders."
Lords of the Flies
Jongens, Penn colleague Balpreet Bhogal, and Brown colleague Robert Reenan studied the fruit fly, Drosophila, whose genome contains a cousin of the human FMR1 gene called dFMR1. The Jongens lab is one of several that use Drosophila models to study Fragile X Syndrome.
"The Drosophila dFMR1 gene is fairly similar to that in humans at the amino acid level - there's about a 50 percent overall similarity between the two proteins," Jongens notes. "But if you look at specific domains, there are pockets of even higher similarity. That makes us fairly confident that some of what we'll discover from the fly model translates to people."
Flies with mutated dFMR1 genes exhibit similar physiological symptoms as humans with Fragile X, including memory and cognitive deficits, disrupted sleep and circadian rhythms, and impaired social behavior.
Two Key Proteins
Another impaired trait due to the mutation is overgrowth and branching of the neuromuscular junction. The research group showed that another key protein critical for normal functioning and architecture at the NMJ is ADAR (dADAR in Drosophila), which is involved in RNA editing of specific mRNAs. The researchers examined the Drosophila NMJ to determine whether dFMR1 and dADAR interact.
They found that the right amounts of the two proteins are important for proper RNA editing and therefore nerve activity at the nerve-muscle synapse. Using a variety of genetic and molecular analyses, Jongens and colleagues were able to link dFMR1 with dADAR via the RNA editing required for normal NMJ structure.
Observing that flies with dADAR mutations displayed similar NMJ defects to flies with dFMR1 mutations, the team demonstrated that both the dFMR1 and dADAR proteins act in the motor neuron to form the neuromuscular junction, indicating that ADAR is required for proper NMJ structure.
In addition, says Jongens, "we found that ADAR acts downstream of dFMR1, suggesting that dFMR1 is required to maintain the proper activity levels of ADAR in the synapse. The FMR protein physically associates with the fly ADAR protein, and through genetic studies we see that there's a dependence of ADAR function on FMRP. If you don't have FMRP, or if you have too much, the editing efficiency of certain sites on select mRNAs is changed. So we think that FMRP might play a role - through the editing process - in modulating the fine tuning of neuronal activity."
The work is the first study to report a disease-associated protein that interacts with and modulates RNA editing. "It provides another way in which the Fragile X Syndrome animal models can be examined to look for defects in neuronal processes that might explain symptoms seen in people," Jongens explains.
Reenan, professor of Molecular Biology, Cell Biology and Biochemistry at Brown, agrees, saying that the implications of the study could be quite broad: "It is a remarkable finding that FMRP can be linked so directly with ADAR and RNA editing activity. Deranged RNA editing has been implicated in epilepsy, suicidal depression, schizophrenia, and even some neurological cancers. These data are surely pointing in the direction of deep connections between numerous distinct diseases of the brain."
The next step in the research will focus on how the two proteins come together to act on the same RNA targets. "The more we know about what the FMR protein does, the more likely we are to uncover ways to treat disease," Jongens points out.