Study finds iPS cells match embryonic stem cells in modeling human disease
(Medical Xpress) -- Stanford University School of Medicine investigators have shown that iPS cells, viewed as a possible alternative to human embryonic stem cells, can mirror the defining defects of a genetic condition — in this instance, Marfan syndrome — as well as embryonic stem cells can. An immediate implication is that iPS cells could be used to examine the molecular aspects of Marfan on a personalized basis. Embryonic stem cells, on the other hand, can’t do this because their genetic contents are those of the donated embryo, not the patient’s.
This proof-of-principle regarding the utility of induced pluripotent stem cells also has more universal significance, as it advances the credibility of an exciting approach that’s been wildly acclaimed by some and viewed through gimlet eyes by others: the prospect of using iPS cells in modeling a broad range of human diseases. These cells, unlike ESCs, are easily obtained from virtually anyone and harbor a genetic background identical to the patient from which they were derived. Moreover, they carry none of the ethical controversy associated with the necessity of destroying embryos.
“Our in vitro findings strongly point to the underlying mechanisms that may explain the clinical manifestations of Marfan syndrome,” said Michael Longaker, MD, professor of surgery and senior author of the study, which was published online Dec. 12 in Proceedings of the National Academy of Sciences. Longaker is the Dean P. and Louise Mitchell Professor in the School of Medicine and co-director of the school’s Institute for Stem Cell Biology and Regenerative Medicine. The study’s first author is Natalina Quarto, PhD, a senior research scientist in Longaker’s laboratory.
Marfan syndrome is an inherited connective-tissue disorder that occurs in one in 10,000 to one in 20,000 individuals. It is caused by any of a large number of defects in one gene. People with this condition tend to be very tall and thin and to suffer from osteopenia, or poor bone mineralization. Medical experts speculate that Abraham Lincoln, for example, suffered from this disorder. Marfan can also profoundly affect the eyes and cardiovascular system.
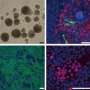
In this study, both iPS cells and embryonic stem cells carrying a mutation that causes Marfan syndrome showed impaired ability to form bone, and all too readily formed cartilage. These aberrations mirror the most prominent clinical manifestation of the disease.
Discovered in 2006, induced pluripotent stem cells, or iPS cells, are derived from fully differentiated tissues such as the skin. Yet they harbor the same capacity of embryonic stem cells to differentiate into all the tissues of the body as well as to replicate for indefinite periods in a dish. Because they offer an ethically uncomplicated alternative to embryonic stem cells, iPS cells have fueled the hope that they can replace ESCs in scientists’ efforts to analyze, in a dish, the cellular defects ultimately responsible for diseases ranging from diabetes to Parkinson’s and even such complex conditions as cardiovascular disease and autism.
One hope for iPS cells is to be able to differentiate them in a dish into tissues of interest — say, nerve cells of a patient with Parkinson’s or autism — and study these resulting cells’ characteristics with an eye to understanding the disease in a patient-specific way. This would be impossible to do with embryonic stem cells, unless ESCs from donated human eggs could be modified through the so-far insurmountable feat of substituting a patient’s own genetic material into these eggs to reflect the patient’s own genetic background.
While scientists have set the goal of using these cells for more than research purposes — developing therapeutic applications in regenerative medicine — that prospect is more distant. Scientists will have to develop the capacity first to repair within such cells, whether iPS or ESC, the genetic defects determined to be responsible for a patient’s condition, and then differentiate the cells in bulk into the affected tissue, which could be used for regenerative medicine. Again, iPS cells in theory might be a better bet because, being initially derived from a particular patient, they could differentiate into tissues that are less likely to provoke graft rejection than similar tissues produced using a donor embryo’s ESCs.
However, a number of studies have reported subtle differences between iPS cells and ESCs, implying that the two may not be equivalent. Experts have wondered whether these differences may render iPS cells inadequate substitutes for ESCs in modeling disease states. This study suggests otherwise, Longaker said.
The opportunity for a head-to-head comparison of ESCs and iPS cells arose serendipitously when Barry Behr, PhD, professor of obstetrics and gynecology and director of the Stanford Fertility & Reproductive Medicine Center, performed pre-implantation genetic diagnosis to select embryos for in vitro fertilization. Behr and Renee Reijo Pera, PhD, professor of obstetrics and gynecology and director of the Stanford Center for Reproductive and Stem Cell Biology, discovered that one candidate embryo carried a genetic mutation that causes Marfan syndrome. This embryo was thus not deemed fit for implantation. But it was a potential source of embryonic stem cells, each of which would carry the Marfan-causing mutation. So, rather than discarding or storing it, the researchers received permission to derive the embryonic stem cells Longaker’s team studied. (Both Behr and Reijo-Pera are co-authors of the study.)
What followed was a collaboration featuring an all-star cast that included senior faculty members from several departments in the medical school as well as researchers at the University of Naples Federico II in Italy. The researchers generated ESCs from the Marfan-carrying embryo. They also obtained skin biopsies from Marfan patients from another Stanford co-author, Uta Francke, MD, professor of genetics and of pediatrics, and used cells called fibroblasts from these samples to derive iPS cells by means of what have now become routine procedures.
“Here we had both iPS cells and embryonic stem cells side by side in culture dishes, both containing the defective gene responsible for Marfan. This was a perfect opportunity to compare them head to head,” Longaker said.
When they did that, Longaker, Quarto and their associates found that both the iPS cells derived from the skin of Marfan patients and the ESCs from the embryonic Marfan carrier exhibited aberrations identical to those that characterize the disorder’s observed skeletal symptoms — a diminished capacity to form bone and a heightened propensity for forming cartilage instead.
The scientists began the study with the knowledge that mutations causing Marfan syndrome are found in a gene that codes for a protein called FIBRILLIN-1. Importantly, FIBRILLIN-1 is known, in turn, to inhibit the activity of an intercellular signaling molecule named TGF-beta. Mouse studies have indicated that the absence or mutation of FIBRILLIN-1 results in a failure of this inhibition. This study showed for the first time in humans that the reason for stem cells’ failure to form bone and overzealous conversion to cartilage directly resulted from their consequent exposure to more, and more-activated, TGF-beta than normal people’s cells are.
The success of iPS cells in faithfully reproducing Marfan’s cellular and molecular defects every bit as well as ESCs do may allow the disease to be studied (and, in the long run, even treated) in a case-by-case manner. While Marfan is a single-gene disorder, it can and does result from any of a large number of mutations to that one gene — upward of 600 have been identified so far —which manifest as a spectrum of subtle differences in symptoms from one patient to the next.