Discovery could increase efficacy of promising cystic fibrosis drug

(Medical Xpress)—A little more than a year after the FDA approved Kalydeco (Vx-770), the first drug of its kind to treat the underlying cause of cystic fibrosis, University of Missouri researchers believe they have found exactly how this drug works and how to improve its effectiveness in the future. Described in the current issue of the Proceedings of the National Academy of Sciences, MU researchers have redefined a key regulatory process in the defective protein responsible for cystic fibrosis that could change the way scientists approach the lethal genetic disease.
"They know the drug works, but they don't know how it works or where it works," said Tzyh-Chang Hwang, PhD, PNAS corresponding author and professor of medical pharmacology and physiology at the MU School of Medicine. "Our paper provides a theory for how Vx-770 works, and based on our understanding of how the CFTR channel works, we have identified a novel strategy for future explorations to complement and enhance the performance of the existing drug."
Cystic fibrosis is the second most common life-shortening inherited disorder occurring in childhood in the United States, after sickle cell anemia. Approximately 30,000 Americans have cystic fibrosis, and there are an estimated 1,000 new cases diagnosed each year. Cystic fibrosis patients are born with a genetic defect that causes a malfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a chloride channel in the cell membrane that plays a critical role in maintaining water and salt balance across many body tissues, such as sweat glands, tissues that line the lungs, liver, pancreas and reproductive organs.
"The chloride channel is like a pipe that allows ions to travel through at a very fast pace," Hwang said. "In cystic fibrosis patients the channel is dysfunctional and activity is diminished. So what is the mechanism that controls the opening and closing of the channel? That is the fundamental discovery of our recent papers summarized in Physiology."
Like an automatic water faucet with a defective hand sensor, many genetic mutations found in cystic fibrosis patients cause a faulty signal, resulting in limited chloride transport across the CFTR. The result is the formation of thick mucus that builds up in the lungs, digestive tract and other parts of the body, which leads to severe respiratory and digestive problems, as well as infections and diabetes.
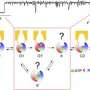
As summarized in Physiology and followed up with further research in the PNAS article, the accidental discovery of a mutation in CFTR, the R532 mutation, allowed MU researchers to reveal a new "non-strict coupling" relationship that occurs between the consumption of ATP, a molecule that provides energy in the body, and the opening and closing of the CFTR. They argue that the new information uncovered about this mechanism that controls the opening and closing of the CFTR and the passage of ions through it could explain how and where the new cystic fibrosis treatment Kalydeco (Vx-770) works.
"To his credit, Dr. Hwang exploited the behavior of the CFTR mutants to demonstrate that CFTR's gate is not strictly coupled to the nucleotide binding engine (NBD) that binds and splits ATP [energy] to drive conformational changes that regulate chloride flow through the CFTR protein channel," said colleague David Sheppard, PhD, an associate professor in the School of Physiology and Pharmacology at the University of Bristol in Bristol, U.K. who did not participate in the study.
In their study, MU researchers were able to observe the effects of the cystic fibrosis drug Vx-770 on the recently discovered R352 mutation. They found that Vx-770 enhances the activity of the CFTR channel by exploiting this "non-coupling" mechanism, a conclusion also supported by experimental results with the wild-type CFTR protein.
"Traditionally, researchers have defined how energy is utilized and transferred in the CFTR as a 'strict coupling' mechanism, meaning that one ATP molecule opens CFTR's gate, ions pass through and the ATP molecule is hydrolyzed and then the gate closes," Hwang said. "In this new model, we argue that the CFTR uses energy from ATP hydrolysis to carry out its function of chloride flow, but this coupling mechanism is more plastic than we thought and therefore could be subjective to manipulations by drugs such as Vx-770."
CFTR is part of a family of thousands of active transporter proteins called ABC proteins. Although CFTR may share many structural features with its ABC "cousins," as Hwang calls them, it has been unclear as to whether CFTR and its cousins may work in a similar manner.
The new idea of how the CFTR utilizes ATP to carry out its function may bear a broader implication because of the evolutionary relationship between CFTR and other ABC transporter proteins. It opens up a wide variety of therapeutic possibilities for other common diseases, such as cancer, heart disease and diabetes, Hwang said, since many other ABC proteins play critical roles in those human illnesses.
"It's taken years for scientists to solve this particular puzzle about the CFTR protein," Hwang said. "Our recent study provides evidence that these ABC transporter proteins and CFTR, a chloride channel, are two peas in a pod. Mother nature employs the same structural framework with just a little bit of modification to do two totally different things. From a basic science perspective, it's a big deal."
Using electrophysiology techniques available at MU's Dalton Cardiovascular Research Center, Hwang's lab studied the opening and closing, or "gating," of the CFTR at the single-molecule level. By measuring the electrical current that reflects directly the movement of chloride ions through one single CFTR channel as it opens and closes, Hwang's lab is able to monitor the activity of a single CFTR molecule in real time.
"Single-channel recording provides a unique opportunity to observe conformational changes in a single CFTR molecule in real time," Sheppard said. "It's exciting to think about how the new models proposed by Dr. Hwang and his colleagues explain the action of Vx-770 and other transformational drugs that target the root cause of cystic fibrosis."
More information: www.pnas.org/content/110/11/4404.abstract