New study points to major discovery for Alzheimer's disease
The Journal of Neuroscience has published a study led by researchers at the Max Planck Florida Institute for Neuroscience, the first and only U.S. extension of the prestigious Max Planck Society, that may hold a stunning breakthrough in the fight to treat Alzheimer's disease. The study potentially identifies a cause of Alzheimer's disease—based on a newly-discovered signaling pathway in cellular models of Alzheimer's disease—and opens the door for new treatments by successfully blocking this pathway. The Institute, which recently opened in December 2012, focuses solely on basic neuroscience research that aims to analyze, map, and decode the human brain—the most important and least understood organ in the body.
"This study transforms our understanding of the direct cause of Alzheimer's disease," said Principal Investigator Dr. Ryohei Yasuda. "With further research, we may open up an entirely new avenue for treatments to combat this disease."
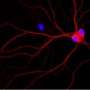
The scientific community so far has widely accepted that Alzheimer's disease is caused by the accumulation of a peptide called Amyloid beta. When Amyloid beta is applied to neurons, neuronal morphology becomes abnormal and synaptic function is impaired. However, how Amyloid beta causes dysfunction is unknown. The MPFI research indicates that the presence of Amyloid beta triggers increased levels of a signaling protein, called centaurin-1 (CentA1), that appears to cause neuronal dysfunction – a potentially groundbreaking discovery that uncovers an important intermediary step in the progression of the disease.
As part of the research, the scientists were able to identify CentA1 and measure its negative effects on neurons. Utilizing an RNA silencing technique, they turned down the cellular production of CentA1, and showed that affected neurons, exposed to Amyloid beta and exhibiting Alzheimer's related symptoms, returned to normal morphology and synaptic function, even with the continued presence of Amyloid beta. They further found that increased CentA1 activates a series of proteins, and these proteins form a signaling pathway from CentA1 to neuronal dysfunction. Thus, inhibiting other proteins in the pathway also "cured" affected neurons.
The initial tests reported were conducted on rat brain slices. MPFI has already started to expand their studies to mouse models of Alzheimer's disease and preliminary experiments show promising results. Ultimately, targeting the components of this newly identified signaling pathway has the potential to open the door for new pharmacological and gene therapies in treatment of Alzheimer's disease. Dr. Yasuda also anecdotally reports that the effects of CentA1 knock down were observed to be sustained over several weeks and an avenue for future study will be to examine how long the positive effects on neurons are sustained which may indicate the potential impact of treatments derived from this research.
More information: The full study will be available at http://www.jneurosci.org/ on March 20, 2013.