Tumor-suppressor protein gives up its secrets, promises new targets for cancer drug design
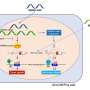
 and inactive (below) forms. Phosphate tags (red) on PTEN's tail cause a shape change, which prevents PTEN from binding to the cell's outer envelope (blue) and removing a phosphate from PIP3 (yellow). Credit: David Bolduc")
Genetic mutations aren't the only thing that can keep a protein called PTEN from doing its tumor-suppressing job. Johns Hopkins researchers have now discovered that four small chemical tags attached (reversibly) to the protein's tail can have the same effect, and they say their finding may offer a novel path for drug design to keep PTEN working.
In a report published on July 9 in the journal eLife, the Johns Hopkins scientists describe how a cluster of four phosphate groups, first found 13 years ago to bind to PTEN's tail, controls its activity.
"Now that we know how these phosphate tags are involved in regulating PTEN's activity, new options may be available for drugs that interfere with them," says Philip A. Cole, M.D., Ph.D., the E.K. Marshall and Thomas H. Maren Professor and director of the Department of Pharmacology and Molecular Sciences at the Johns Hopkins University School of Medicine.
In addition to phosphate tags, mutations in genes that code for a protein can cause some protein activities to be permanently on or off. Mutations that deactivate PTEN often lead to cancer because PTEN's job is to prevent cells from dividing too much, Cole notes. But there are times when a cell needs to divide to replace dead cells, so scientists knew there had to be a naturally occurring mechanism for turning PTEN off, and they hypothesized that the phosphates on its tail were responsible, as they are in other proteins.
To get at the heart of the question, the Johns Hopkins team had to overcome technical obstacles, including a way to engineer a special version of PTEN in which the phosphates were permanently bound to the tail.
"We had to synthesize the tail of PTEN in the lab and then fit that together with the rest of PTEN, which was made by insect cells," says David Bolduc, a graduate student in Cole's laboratory and the lead author of the paper. "Once we cleared that hurdle, we were able to learn a lot more about how phosphates regulate PTEN."
Armed with their engineered protein, the team analyzed its shape, where in cells it was located and its activity—tasks aided by miniature X-ray imagers and biochemical tests that shed light on how PTEN interacted with other entities, like PIP3, a fat-like molecule located just inside the outer envelope of cells.
The team found that when there are no phosphates on PTEN's tail, it is in its active form and it removes a phosphate tag from PIP3. The loss of the phosphate alters PIP3's activity and causes a chain reaction of effects on other important regulatory proteins that ultimately suppresses cell division and migration, both deadly aspects of tumor progression.
Cole explains that when a cell needs to divide, another protein, most often CK2, adds phosphates to PTEN's tail, causing a change in its shape and location. Its tail curls back on the rest of the protein and prevents it from interacting with PIP3 in the outer envelope of the cell, so PTEN ends up inactive, in the fluid-filled middle of the cell. When the phosphates are removed, PTEN relocates to the outer envelope, where it removes a phosphate tag from PIP3 to initiate the chain reaction that suppresses tumor formation.
"The tail of PTEN actually has a competition going on between binding to itself and binding to the outer envelope of the cell where PIP3 is located," explains Bolduc. "Any drug that can prevent the tail from binding itself might also maintain the tumor-fighting activity of PTEN."
According to Cole, many cancer patients have an overabundance of CK2, the protein that adds phosphates to PTEN and turns it off. So, increasing the activity of PTEN might be helpful not only to patients with defective PTEN, but also to those with cancer-causing mutations in other proteins.
More information: dx.doi.org/10.7554/elife.00691