A predictive fitness model for influenza
Researchers at Columbia University and the University of Cologne have created a new model to successfully predict the evolution of the influenza virus from one year to the next. This advance in our understanding of influenza suggests a new, systematic way to select influenza vaccine strains. The findings appear in Nature on Feb. 26.
The flu is one of the major infectious diseases in humans. Seasonal strains of the influenza A virus account for about half a million deaths per year. In a concerted effort, WHO and its Collaborating Centers have closely monitored the evolution of the seasonal H3N2 influenza strains for over 60 years. Based on these data, influenza strains are selected for vaccine production twice per year. Because influenza is a fast-evolving pathogen, the selection of optimal vaccines is a challenging global health issue.
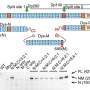
In recent years, it became clear that the evolution of the flu is a complex process. Different influenza strains compete with each other; the race is about how to successfully infect humans. This prompted Marta Łuksza, of Columbia's Biological Sciences department and Michael Lässig of the Institute for Theoretical Physics at the University of Cologne, to ask the question: Can we predict which of these competitors will win the race? "This was a challenge for an evolutionary biologist because there are very few systems in the wild for which quantitative predictions of their evolution are at all feasible," says Łuksza. "It was also a computational and theoretical challenge. While traditional evolutionary thinking is about reconstruction of the past, we had to develop ideas on how to reach into the future." Most importantly, the scientists had to find out which part of the system can be actually predicted and which are random. In their approach they used ideas from physics and computer science.
Łuksza and Lässig used Darwin's principle: survival of the fittest. But what determines how fit an influenza virus is? First, they considered innovation: the virus had to keep a high rate of mutations in order to escape from human immune response. But they also included conservation: these mutations must not compromise the essential functions of a virus, such as the correct folding of its proteins. Through studying the genomes of the virus, they devised a way to predict which viral strains have the optimal combination of innovation and conservation.
While Łuksza and Lässig focused on influenza, their approach highlights a general link between evolution and its consequences for epidemiology that is relevant for many fast-evolving pathogens. In a broader context, it touches upon the fundamental question of how predictable biological evolution is. "There is clearly no general answer to this question," says Łuksza. "But our analysis shows under what auspices limited predictions may be successful." Further extensive tests with global influenza data would help determine whether their method would lead to improved vaccines.
More information: Nature DOI: 10.1038/nature13087